
While the majority of the first workshop focused on exploring the future potential of real-world data (RWD) and real-world evidence (RWE), and identifying the challenges that need to be addressed, this final session instead focused on dissecting the current system of evidence generation. Evidence for decision making, particularly regulatory decision making, is traditionally generated through randomized controlled trials (RCTs). The first part of this chapter explores the drawbacks and misconceptions about RCTs and other current methods, while the second part of the chapter discusses ways in which the system could be improved, including through the incorporation of RWD and RWE.
FROM PRECISION TO RELIABILITY
The traditional system of evidence generation, said Robert Califf, has done enormous good. It has delivered evaluations of the benefits and risks of medical products and interventions that have enabled these technologies to have a dramatic impact on life expectancy, physical function, and the ability to enjoy life. However, the traditional evidence-generation system has become “bloated and burdened” with practices that massively increase the cost of research without necessarily improving the quality, he said. Califf said that while the old system has not failed, the current time is an important inflection point with the opportunity to refocus efforts and dramatically accelerate the generation of evidence while also improving quality.
Science is in an “explosive phase,” said Califf, in which there is a proliferation of technologies and new approaches for research, prevention, diagnosis, and treatment. At the same time, the costs of doing research are increasing. In some cases, this has resulted in “putting things on the shelf because we can’t afford to do the development,” said Califf. In addition, the cost of health care has been rising, which is leading health systems to try to assess the comparative value of old and new therapies. The result of all of these changes is a dramatic need to generate more high-quality evidence about diagnostic and therapeutic technologies and clinical strategies. However, the current evidence-generation system is well intentioned, but flawed: it is expensive, slow, not always reliable, unattractive to clini-
cians and administrators, and does not answer the questions that matter most to patients. This old, unsustainable system, said Califf, was built at a time when technology was limited, clinical notes in health records were handwritten, and there were few electronic data in the context of routine clinical care. As computing advanced faster in non-medical sectors than in the practice of medicine, research experts developed parallel systems to record clinical findings entirely separately from clinical practice; the perpetuation of this “parallel universe” of data and antiquated systems led to some “bizarre” inefficiencies. For example, said Califf, as electronic health records (EHRs) developed, research coordinators were instructed to print out notes in order to produce a written record, which would then be checked against the electronic system. These types of “arcane practices” were codified and amplified through the development of good clinical practices (GCPs) and standard operating procedures (SOPs), he said.
One practice that is particularly troublesome, said Califf, is the idea that recording each data point with as much precision as possible will result in a more reliable estimate of treatment effect. However, this “patently incorrect” belief results in wasting millions of dollars, without an appreciable increase in the quality or utility of the evidence generated. As an example, Califf pointed to a case in which thousands of patients were studied to determine the dosing regimen of a certain drug. A U.S. Food and Drug Administration (FDA) inspector expressed a lack of confidence in the results because the exact time the drug was ingested was not recorded. Recording the time of ingestion would have cost “probably on the order of $10 million,” and would have likely contributed little information in a drug administered twice per day. What is needed now, said Califf, is moving away from this narrow focus on precision to a broader focus on reliability. Clinical trials should be designed and conducted in order to produce reliable results that meet the needs of patients, providers, payers, and policy makers, said Califf.
Califf drew a distinction between a system focused on precision and a system focused on reliability by offering the definitions of each word:
- Precision: (1) The quality, condition, or fact of being exact and accurate. (2) Refinement in a measurement, calculation, or specification, especially as represented by the number of digits given.
- Reliable: Consistently good in quality or performance; able to be trusted.
Califf explained that for some types of research, precision is critically important. For example, if small samples and measurements are expensive to take, precision is essential. Precision is also important in early phases of research when little is known, or for research where the administration
of a drug must be carefully timed. However, for many studies, a focus on precision limits the potential of research by creating budget requirements that severely limit the size of the study that can be conducted, the duration of follow-up, or the number of endpoints that can be examined. For these studies, the more important characteristic is that the results are dependable, sound, and able to be trusted, he said. In essence, these studies should focus on “providing the answers to the questions that matter to the patients.”
An evidence-generating system that focused on reliability, said Califf, would have four key principles:
- Build a reusable system embedded in practice;
- Use quality by design;
- Use automation for repetitive tasks; and
- Operate from basic principles.
First, an evidence-generating system focused on reliability would be a learning system that is embedded in clinical practice, and would enable learning from every encounter (see Figure 5-1). This is an old concept in health care and is a fundamental concept in business. In addition, the lessons learned would return to the point of care and be used to improve care. Califf observed that this capability is being developed by public–private partnerships and integrated health systems, and noted that “if these various systems can work together in a federated way, I think we are getting close to having a national system that can be reused at a very low cost for different kinds of questions.” This vision of a new national system would collect data during routine care, would use active surveillance to protect patients, would leverage RWE to support regulatory decisions, and could be used to inform decisions by all stakeholders in the ecosystem, said Califf.
Second, the system would use a quality-by-design approach in order to focus on and eliminate errors that bias the results, while ignoring errors that do not affect the outcome. Trying to eliminate all errors, said Califf, is costly, inefficient, and unnecessary. Califf said the quality-by-design process requires researchers to think through the objectives of the trial, identify the factors that are critical to meeting those objectives, and work to mitigate the risks that are likely to lead to errors that matter. Califf directed workshop participants to the quality-by-design toolkit for further information.1
The third principle of a reliability-focused system, said Califf, would be to capitalize on the rapidly expanding technologies and infrastructure that are available. Examples would be using automation for repetitive tasks, performing real-time analysis of data that are routinely collected, and
___________________
1 The Clinical Trials Transformation Initiative’s quality-by-design toolkit can be found at http://www.ctti-clinicaltrials.org/toolkit/QbD (accessed November 2, 2018).
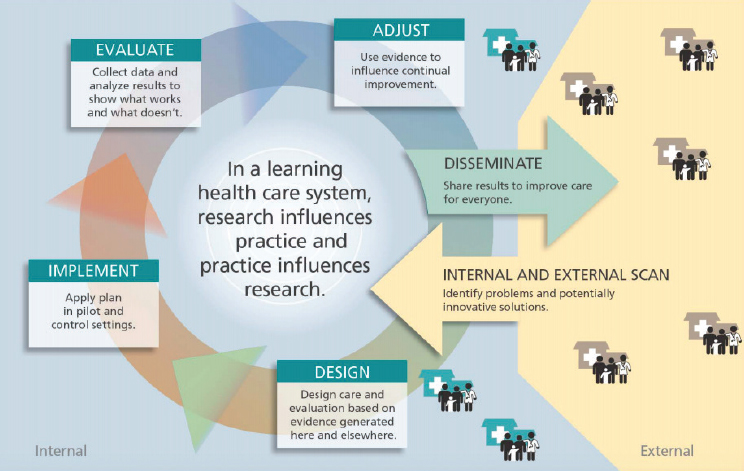
SOURCES: Califf presentation, September 20, 2017; Greene et al., 2012.
developing infrastructure to share the results with practitioners to support a constantly learning system. Califf noted that automated analysis of data could help fill in the evidence gaps on endpoints that are less well understood, at a considerably lower cost.
Finally, a reliability-focused system should operate from basic principles of good scientific research, rather than simply creating another “bureaucratic entanglement” of new and different SOPs (see Box 5-1). Some basic principles, said Califf, would include
- Focusing on errors that matter;
- Enrolling study participants who are likely to inform the question;
- Randomizing;
- Masking;
- Measuring outcomes in a manner that is fit for purpose;
- Considering strengths of different designs for different purposes; and
- Designing operations that yield an answer to the question in an efficient manner.
INTEGRATING THE NEW WITH THE OLD
The traditional evidence-generation system, said Califf, is not necessarily broken, but needs dramatic improvement. Several speakers addressed the issue of how to integrate RWD and RWE with the traditional evidence-generation system, and more generally, how to generate better answers for questions, no matter what method or source of data.
Real-World Evidence to Address Challenges Along the Drug Development Pathway
John Graham, head of value, evidence, and outcomes at GlaxoSmithKline (GSK), echoed other speakers in his opening remarks: “We need to have the right answers to the right questions at the right time.” In getting these answers, said Graham, RWE is a must-have, but it is not a replacement for traditional research. Using both traditional and new methods of research
and sources of data will lead to a richer and more informative body of knowledge to inform decisions. GSK has been trying to move from a “study-by-study” assessment process to a “challenge-based thinking process,” said Graham. The challenge-based process starts with clarifying the end goal: What does the patient need to have an improved outcome? The second step is identifying the challenges to getting the patient to that improved outcome. Finally, “We look for a book of work that can resolve that challenge,” he said. The book of work can include multiple sources of data and types of studies, and can include evidence from traditional RCTs as well as RWE. Graham stressed that trials that include randomization are essential for understanding causation and laying a base of knowledge. However, RWE can be a useful adjunct to RCTs in order to expand understanding of disease and the patient experience.
Graham said that there are broad uses for RWE along the drug discovery and development pathway (see Figure 5-2). Much of the focus is often on using RWE in prediscovery or in postmarketing evaluation. However, said Graham, there are unique capabilities of RWE that can be used in other phases as well. For example, social media can be used to understand the perspectives and needs of patients. Graham noted that patient advocacy groups often give the perspective of the “professional patient,” but social media allows GSK to tap into the “real-world” patients. He said that as researchers are planning RCTs, they can design components such as outcomes, measures, and tools in ways that are most appropriate for the patients. Another unique use of RWE is to understand the thresholds of effect that are most important for patients; for example, a drug could be produced that results in a small reduction in blood glucose in diabetes patients, but this effect might be too small for patients to want to take the drug.
Each step of drug discovery and development has challenges and tough decisions; RWE can help focus drug development along the entire pathway, said Graham. RWE can be used during discovery to estimate unmet needs, or to characterize patient heterogeneity. RWE can be used during development to form clinical pathways, optimize trial design, and optimize price. RWE can be used after approval to study comparative effectiveness, learn about compliance and adherence patterns, and investigate effectiveness in subpopulations. RWE, concluded Graham, is not about one method or one source of data, but about using information to overcome challenges and improve patient outcomes.
The Need to Streamline and Continue the Use of Randomized Controlled Trials
Several speakers at the workshop identified problems with the traditional reliance on RCTs, and proposed using non-randomized observational
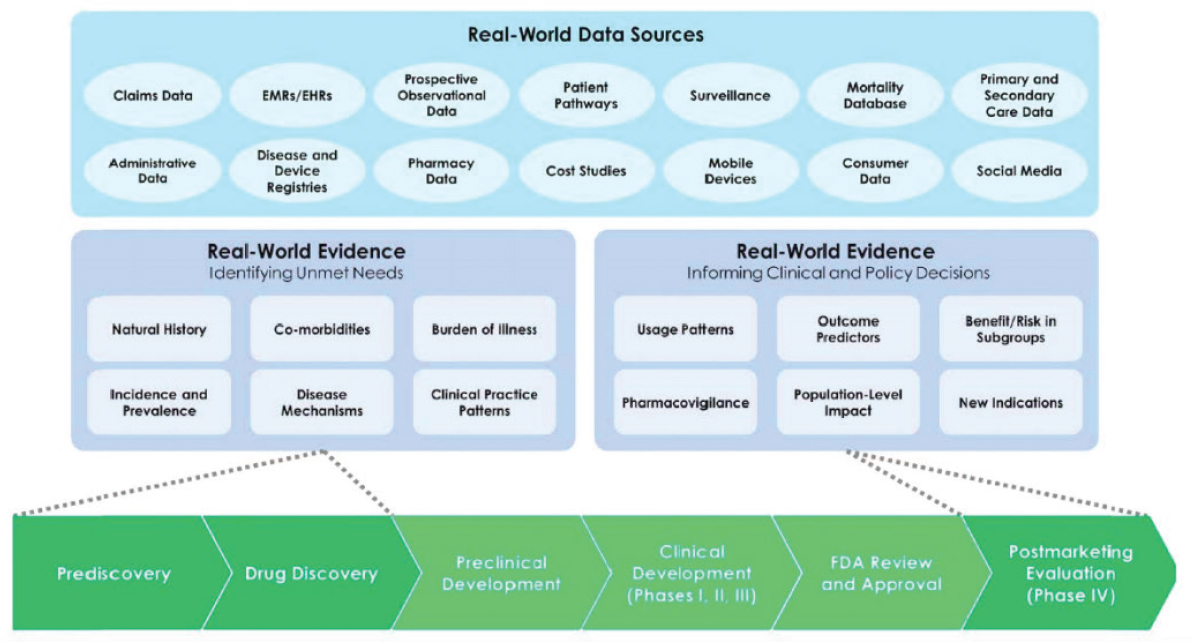
NOTE: EHR = electronic health record; EMR = electronic medical record; FDA = U.S. Food and Drug Administration.
SOURCES: Graham presentation, September 20, 2017; Galson and Simon, 2016.
studies of RWD as an alternative to RCTs. Rory Collins presented a different perspective: RCTs are overly regulated, unnecessarily expensive, and focused on rules that are not based on scientific principles. In his opinion, the solution is not to replace RCTs with non-randomized observational studies, but instead to make it easier to do RCTs.
While non-randomized observational studies may be useful for detecting large effects of treatments on health outcomes that are rare, RCTs are necessary for detecting moderate effects of treatments on common health outcomes reliably, said Collins. Non-randomized observational studies, he said, are limited in detecting moderate treatment effects and causal associations. When based on large databases, such studies may find associations of health outcomes with treatments that are highly statistically significant and precise, but that does not mean they are causally related, Collins said. This is because the underlying risk of people who take the treatment and those who do not may differ systematically, even after statistical adjustment. By contrast, randomization allows differences in outcomes to be causally attributed to treatment, because the randomized patient groups differ only randomly from each other in terms of their underlying risk of events. Randomization also allows use of a blinded control group, which can help ensure events are ascertained similarly in the randomized treatment groups, yielding unbiased treatment comparisons.
The current challenges with RCTs, said Collins, are in large part due to the widespread misapplication of the GCP guidelines for clinical trials issued by the International Conference on Harmonisation (ICH). Collins said these guidelines are not based on key scientific principles that are critical for the generation of reliable results in RCTs, and that the complexity and costs of adhering to them are unsustainable. In addition, ICH-GCP is applied far more widely than its original purpose: It was developed only for registration trials of new drugs, but compliance with it is now also required by governments (e.g., the European Union Regulation for Clinical Trials) and non-commercial funders (e.g., the Gates Foundation). Collins gave several examples of ICH-GCP–related practices that are wasteful, inefficient, and ineffective:
- Requirement to record all adverse events (AEs), not just serious AEs;
- Requirement to record narratives for all serious AEs in case there is an excess of a particular AE;
- Demands for unblinded results for AEs (including even primary outcomes) during ongoing trials; and
- Annual reports required by regulatory authorities that are so long that safety signals risk being lost.
In short, the ICH-GCP guidelines put undue emphasis on the quality of the data in RCTs, said Collins, rather than on the generation by RCTs of reliable results about the safety and efficacy of the treatment being studied. In his opinion, focus on compliance with rules due to overregulation and related bureaucracy, rather than on innovative designs and good results in RCTs, has resulted in obstacles, delays, and high costs. As a consequence, he contended, it has led some researchers to pursue the alternative of using non-randomized observational studies—what he called the misuse of RWE—to assess treatments, despite their potential for biases. Noting that the ICH-GCP guidelines require specific qualifications for investors, source data verification, and regulatory documentation, Collins argued there is an urgent need to improve RCT methodology through the development of comprehensive new RCT guidelines based on key scientific principles required to generate evidence about the safety and efficacy of treatments that can be trusted.
Other individual workshop participants noted that a purpose of ICH-GCP was to give providers guidelines for how to conduct research well. Janet Woodcock, director of FDA’s Center for Drug Evaluation and Research, noted that regulations are designed to protect people, as well as the scientific enterprise, from bad actors. However, with this caveat, Woodcock said that regulations for the 21st century need to have a more “flexible understanding of quality” and a structure that allows for consistent monitoring and course correction, rather than rigid and unyielding rules.
From “One Study at a Time” to “All by All” Analyses
Patrick Ryan, senior director and head of epidemiology analytics at Janssen Research & Development, spoke to workshop participants about the Observational Health Data Sciences and Informatics (OHDSI) program. OHDSI is an open science community, said Ryan, where anyone can participate in conducting research on observational databases. The goal of OHDSI is to “improve health by empowering the community to collaboratively generate evidence,” he said. OHDSI operates across 20 different countries, with more than 200 researchers. Similar to Sentinel, OHDSI has a distributed data network with open community standards. Collectively, there are more than 60 databases that contain patient records for 660 million patients. OHDSI’s strategy, said Ryan, includes methodological research in order to establish and evaluate scientific best practices before applying them to observational data. The results of this research are codified into open-source tools that the entire community can use, with all code shared on GitHub (a Web-based repository for code).2 This open-source approach,
___________________
2 For more information about GitHub, see https://github.com (accessed November 2, 2018).
said Ryan, is part of OHDSI’s “moral obligation to . . . generate the evidence and get it out to patients as quickly as possible.” Ryan described the three focal points of OHDSI’s research:
- Clinical characterization: The diagnoses, treatments, and outcomes for a population;
- Patient-level predictions: The probability of an individual patient developing the disease or experiencing an outcome; and
- Population-level effect estimation: What are the causal effects between treatments and outcomes?
Focusing on population-level effect estimation, Ryan conducted a live demonstration of evidence analysis for the workshop participants. Ryan started with a paper about antidepressant medication use and the risk of preeclampsia in pregnant women with depression (Avalos et al., 2015). The study found an observed association between antidepressants and preeclampsia, and found that the association was stronger for selective serotonin reuptake inhibitors (SSRIs) in particular, with a statistically significant relative risk of 1.4, said Ryan. Another observational study, said Ryan, looked at the same question, using data from the Medicaid population (Palmsten et al., 2013). This study, in contrast with the first, found that other types of depression medications (serotonin and norepinephrine reuptake inhibitors and tricyclics) were associated with a higher risk of preeclampsia than SSRIs. In this study, SSRIs had a non-statistically significant relative risk of 1.00. A third paper that Ryan presented was a meta-analysis of research that looked at the link between antidepressants and preeclampsia. This meta-analysis concluded that “while some studies have suggested a moderately increased risk, the current data do not allow for a definitive conclusion.” The meta-analysis pointed out the methodological limitations of many of the studies, and the fact that untreated depression and anxiety could not be disentangled from the results.
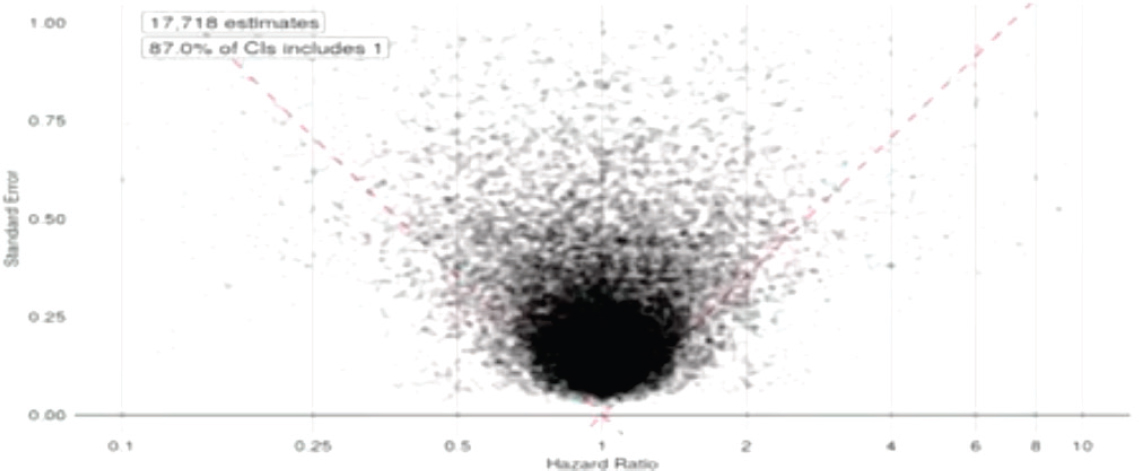
Ryan showed the audience a funnel plot that contained data from these studies, as well as other studies that had been mined from the published literature on the topic of antidepressants and preeclampsia (see Figure 5-3). The pattern on the funnel plot, said Ryan, “should be alarming.” The plot showed that the evidence is skewed toward the right, that is, more results are positive than negative. In addition, 70 percent of the results are statistically significant and many of these are hovering right at the dashed line that represents a p-value3 of 0.05. This pattern, said Ryan, suggests that “some-
___________________
3 A p-value represents the probability of finding the observed results if the null hypothesis were true. A p-value of less than 0.05 is often used to determine whether a result is statistically significant.
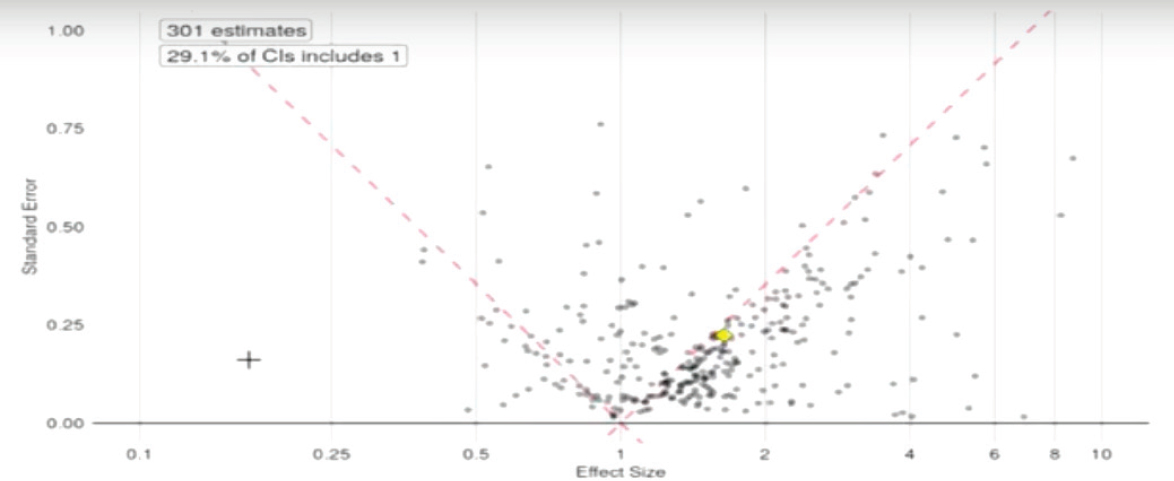
NOTE: CI = confidence interval.
SOURCE: Ryan presentation, September 20, 2017.
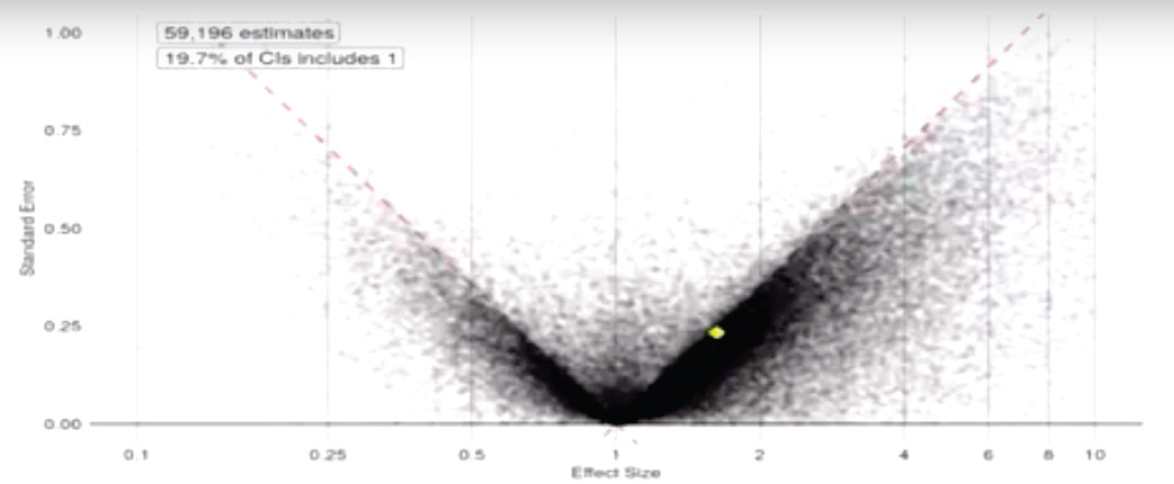
one kept working at the data until they got p less than 0.05 and then quit.” Ryan showed the workshop participants another funnel plot that plotted 60,000 published observational studies on multiple disease states (see Figure 5-4). This plot showed that, again, the studies were skewed toward positive results, and 80 percent of the published studies were statistically significant, with many studies hovering right at the 0.05 line.
This exercise, said Ryan, demonstrates that “we can’t necessarily trust the process that we are using to generate evidence as a community.” Our current process, Ryan said, is to conduct one observational study at a time, with one hypothesis, one dataset, and one method. Each of these studies is viewed individually, but given the pattern on the funnel plot, “it can’t possibly be the case that all of these studies are totally correct.” The process of generating evidence in RCTs, said Ryan, is not much better. He pointed to a meta-analysis of multiple clinical trials on depression. The meta-analysis concluded that there was not sufficient evidence to draw conclusions about the comparative risk of side effects, including suicidality, cardiovascular events, and seizures. If “we still don’t know the answer, despite decades of research and hundreds of millions spent on this question,” he asked, what could be done differently?
Ryan suggested that obtaining the answers we need is best done by considering the patient perspective. An individual patient with depression, said Ryan, wants to know which of many available treatments would be best for him or her. Different patients may prioritize different factors; for example, one patient may want to know about suicidality while another is more concerned about hepatotoxicity. Ideally, every patient would have access to every personally relevant data point. The way to do this, said Ryan, is through an observational data network, like OHDSI, of multiple standardized data sources that can answer questions one at a time. For example, a person could “ask” the network about whether one specific antidepressant increases the risk of diarrhea more than another. The observational data in the network may or may not be statistically significant, and may have large or small effect estimates, but the person asking the question can see all of this information and make a decision accordingly.
The question that remains, however, is how do researchers know these data are reliable? Ryan showed workshop participants another funnel plot. This figure graphed every observational data point within the OHDSI system, comparing all antidepressants against each other on all outcomes—an “all by all analysis.” This funnel plot (see Figure 5-5), unlike the plots from RCTs, does not have a preponderance of data hovering around the line of statistical significance, and the huge majority of the dots are not statistically significant. Because these data are not subject to a researcher or a publication deciding what to publish, they reflect the true breadth of data and not a subjective selection. This pattern, said Ryan, suggests that the evidence
NOTE: CI = confidence interval.
SOURCE: Ryan presentation, September 20, 2017.
NOTE: CI = confidence interval.
SOURCE: Ryan presentation, September 20, 2017.
in these databases is less biased than the evidence from the published trials, and therefore more reliable for making decisions. There is still variability within these data, said Ryan; however, the systematic approach helps to remove the variability that is introduced and leaves the variability that is inherent to patient heterogeneity and health system bias and other factors.
Randomized trials, said Ryan, are still appropriate for many purposes. However, in the current system, there are many clinical areas in which trials have not occurred and practitioners operate without evidence. Using this systematic approach to examine the totality of observational evidence in order to generate answers is one solution to this evidence gap, he said.
REGULATORY PERSPECTIVE
Woodcock agreed with the speakers on several points. The current evidence-generation system for medical products is very costly and time consuming, and leaves many questions about product use unanswered. One consequence of this situation, Woodcock said, is that many clinical decisions are not evidence based because generating the answers is too expensive. As one potential solution to this problem, Woodcock said that “FDA is committed to exploring the use of real-world evidence in regulatory decisions.”
FDA is exploring the use of RWE in several ways, Woodcock said. In the drug space, FDA is involved in a demonstration project called IMPACT-AFib, a randomized educational intervention that uses Sentinel to examine outcomes (see Richard Platt’s presentation in Chapter 3). In the device space, FDA’s Center for Devices and Radiological Health and Center for Biologics Evaluation and Research issued guidance in mid-2017 about the use of RWE for device decisions. The device guidance, titled “Use of Real-World Evidence to Support Regulatory Decision-Making,”4 discusses the challenges with current device evidence development, and proposes potential uses of RWE for device regulation. These uses include using RWE to examine outcomes, but also as historical or concurrent controls, to expand the label for an approved device, or for safety surveillance. This guidance, said Woodcock, should serve as an incentive for the device industry to invest in making RWE generation more robust. Similar considerations apply to the use of RWE for drug approval, said Woodcock. RWE has long been used for the evaluation of safety in the postmarketing of products, but there is little historical use of RWE for decisions about effectiveness. However, she noted that “there are no hard and fast rules” about how evidence must be generated for drug approval, with the exception of rules about
___________________
4 See https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm513027.pdf (accessed November 7, 2018).
informed consent and patient privacy. FDA is open to a wide spectrum of evidence, from standard clinical trials to pragmatic trials conducted in the health care system. However, she noted, there are trade-offs involved in the choice of trial design, including data reliability, pragmatism, control of errors, safety, and other factors, and FDA would consider these trade-offs when evaluating RWE. For example, it would be inappropriate to run a first-in-human trial in a real-world setting, she said.
Woodcock gave several examples of ways in which FDA has used or is considering using RWE for drug approval. Drugs for rare diseases have been approved using data from registry-like case series, she said. For example, Lumizyme for Pompe disease was approved using survival data from an international registry of infantile-onset disease. Registry data have also been used for external controls for uncontrolled experience data, she said. FDA is exploring how randomization would work in registry or health care settings, and they are collaborating with other stakeholders to improve the validity of key data elements that are collected during the course of health care. Woodcock referred to the quality-by-design approach that Califf had mentioned, and said using this approach ensures that “you get it right the first time”—data are put into the EHR correctly and do not have to be adjudicated and curated later.
There are several potential uses for RWE during the drug development process, said Woodcock, including
- Natural history information: RWE is valuable for learning about patients’ experiences with a disease, and what their burdens and needs are. RWE can help develop appropriate outcomes for a study, based on patient progression and self-reported outcomes. This is particularly true for rare diseases and/or diseases that are very heterogeneous. Rare disease experts, she said, are often wrong, because their opinions are based on the few patients that they have seen, so it is essential to get RWE from as many patients as possible.
- Biomarker development: Understanding biomarkers and choosing appropriate markers is important for developing drugs in an efficient way. Biomarkers that are critical to development should be explored in humans as thoroughly as possible before initiating a study, and RWE approaches could be essential to gathering this information.
- Hybrid model for investigational drugs: There are ways to combine traditional study approaches with RWE. For example, an investigational drug could be evaluated in a hybrid model that uses traditional randomization for initial assignment of patients and uses RWE to measure outcomes. This approach requires integrat-
-
ing the trial into the health care process, and collaborating with caregivers as research partners. A good example of this approach is the National Institutes of Health Collaboratory.
- Add new indications to an approved drug: When extending the label of an approved drug, RWE may be compelling enough that an RCT is not needed. For example, ivacaftor, a drug used for cystic fibrosis, could have been approved for additional mutations based on registry data, combined with trial and mechanistic data.
Woodcock said that for any use of RWE, the important part is ensuring that the research protocol is designed thoughtfully and appropriately. If the design is excellent and takes into consideration potential errors and how to manage them, FDA or any other regulatory body could agree on the alternative design and agree to accept the evidence.
One design approach that is particularly promising, said Woodcock, is the use of master protocols. Master protocols are continuous, ongoing trials that can study multiple interventions and outcomes, with the goal of having “continuous improvement in the disease outcome.” The use of master protocols, said Woodcock, saves time, offers an opportunity to include community practitioners and integrate research and practice, can answer multiple questions, is patient-centric, and can use adaptive designs creatively. However, there are also challenges involved with this approach: It is a novel approach that is difficult to set up at the beginning, and it does not comport with the traditional models of pharmaceutical development, academic rewards, or grant funding. Master protocols offer an opportunity to incorporate RWD as extensively as possible, said Woodcock, although this will require additional work in standardization, data verification, training, and curation. These initial investments, however, will likely pay off in terms of lower costs, greater efficiency, the engagement of first-line practitioners, and the ability to answer more questions.
Science and medical care are rapidly changing, said Woodcock, and these changes mean “that we are going to have to change our traditions.” More rare and orphan diseases are being studied, and even in common diseases, there are targeted therapies with companion diagnostics. These changes are narrowing the target population for a medical product in such a way that traditional trials do not work very well, she said. The current inability to efficiently generate needed evidence for drug development and for clinical practice, she said, will continue to be a major barrier to innovation and the quality of care. Drug developers and regulators will have to adapt to this new world with innovative designs and the use of RWE to get the answers that patients need. Woodcock stressed the need for pragmatism in research and for improving the current situation. She noted that what clinicians do now is often “based on observational studies or, even worse,
individual experience.” The research community could instead focus on making evidence generation easier and more efficient, while still emphasizing reliability, in order to get the answers that patients, providers, and regulators need to make decisions.
This page intentionally left blank.




















