
Obscuring intervention allocation—commonly known as blinding—is used to reduce the likelihood that those involved in the trial will be influenced by the treatment assignment, said James P. Smith, deputy director of the Division of Metabolism and Endocrinology Products at the U.S. Food and Drug Administration’s Center for Drug Evaluation and Research. If those involved are influenced by treatment assignment, it may affect the outcome of the trial. Blinding allows researchers to study the effects of the intervention without the influence of patient and provider perceptions. However, blinding is not always appropriate or feasible, said Alex John London. Trial design features such as blinding have to be justified by the contribution they make to the evidence quality and the relative risks and costs compared with alternative designs, he said. This session in the third workshop examined the topic of blinding, and session moderator Jonathan Watanabe, associate professor of clinical pharmacy and National Academy of Medicine Anniversary Fellow in Pharmacy, University of California, San Diego, asked participants to consider the following:
- How might variability in knowledge of treatment group assignment affect provider and patient adherence and outcomes?
- How might variability in knowledge of treatment assignment affect study cost and reliability?
- What key factors could affect decisions to obscure intervention allocation?
This topic was highlighted by some participants at the second workshop as an area that needed further exploration, so a session was held on the topic at the third workshop.
ILLUSTRATIVE EXAMPLES
To explore the issues surrounding obscuring treatment intervention and blinding, speakers at the second and third workshops presented case studies as illustrative examples of the considerations that go into designing and conducting a real-world study.
INVESTED Trial
Orly Vardeny, associate professor of medicine at the University of Minnesota and of the Minneapolis VA (U.S. Department of Veterans Affairs) Center for Chronic Disease Outcomes Research, told workshop participants about the INVESTED (INfluenza Vaccine to Effectively Stop Cardio Thoracic Events and Decompensated heart failure) trial. Influenza leads to significant morbidity and mortality, said Vardeny, and there have been several analyses that documented a temporal association between influenza infection and cardiovascular events (Madjid et al., 2007; Thompson et al., 2003, 2004). Researchers have sought to further understand how the influenza vaccination could affect cardiovascular events. The first step, said Vardeny, was a meta-analysis of six studies that demonstrated that influenza vaccination could reduce cardiovascular events (Udell et al., 2013). The next research question, said Vardeny, was to determine if patients with heart failure exhibited the same immune response to the flu vaccine. This analysis showed that heart failure patients had a reduced antibody response compared with patients without heart failure (Vardeny et al., 2009). Researchers were able to increase the antibody response by giving a higher dose of the flu vaccine, she said (Van Ermen et al., 2013).
Based on this initial research, the INVESTED1 researchers set out to examine whether the increased immune response from the high-dose vaccine would translate into better cardiovascular outcomes (see Figure 8-1). The researchers designed a randomized trial comparing high-dose trivalent influenza vaccine to standard-dose quadrivalent influenza vaccine. Vardeny noted the standard-dose quadrivalent vaccine for the active control group was chosen because it would be unethical to have an unvaccinated control group, and the quadrivalent vaccine is the current standard of care. Researchers have enrolled approximately 3,000 participants out of a target of 9,300 participants for this pragmatic study so far, and have contacted them up to four times per year by phone to ascertain endpoints. The primary endpoint is a composite of death or cardiopulmonary hospitalization.
Both providers and participants are blinded in the study, which is accomplished by a third-party vendor affixing identical labels over the individual-dose syringes. Vardeny said that because there were some ways to tell the two vaccines apart (e.g., the high dose causes more pain during injection), the study was designed so that the person who administers the vaccine is not the same person who conducts the ascertainment phone calls later in the season.
Vardeny said the choice to conduct a double-blind study was based on a few considerations (see Box 8-1). There were reasons not to blind,
___________________
1 See https://clinicaltrials.gov/ct2/show/NCT02787044 (accessed January 4, 2019).
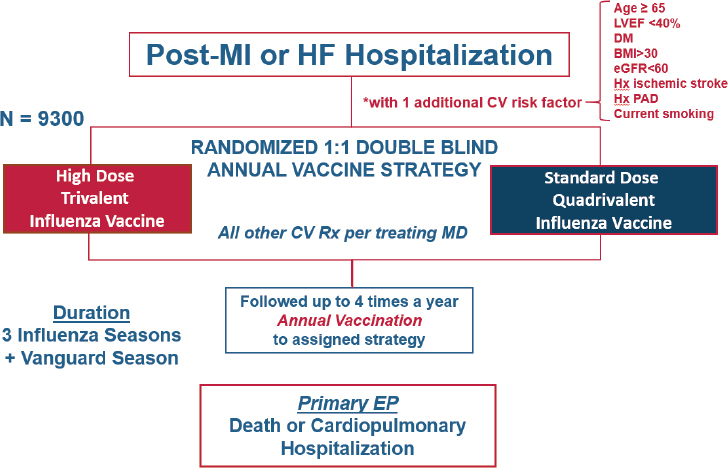
NOTE: BMI = body mass index; CV = cardiovascular; DM = diabetes mellitus; eGFR = estimated glomerular filtration rate; EP = end point; HF = heart failure; Hx = history; Hx PAD = history of peripheral artery disease; LVEF = left ventricular ejection fraction; MD = Doctor of Medicine; MI = myocardial infarction; Rx = prescription.
SOURCE: Vardeny presentation, July 17, 2018.
she said. Blinding delays the study by several weeks while the third-party vendor blinds and distributes the vaccines. During this period, the flu vaccine becomes available to the general public, which reduces the number of people eligible to participate in the trial because they already received the vaccine. However, said Vardeny, the reasons for blinding outweighed these considerations. There are perceived differences in efficacy of these vaccines, she said. For example, the high-dose vaccine is only approved for individuals aged 65 and over. Although there is no recommendation that the standard of care for people over 65 should be the high-dose vaccine, some providers or patients may perceive that all people over 65 should get the high-dose vaccine. Without double blinding, these perceptions may have resulted in systematic bias in terms of patients presenting at the hospital, or providers urging hospitalization.
DECISION AID
The general issues discussed by individual workshop participants in the first and second workshops were used to develop a decision aid for the third workshop (see Figure 8-2). As with the other decision aids, the intention was to outline some questions to consider in order to make thoughtful
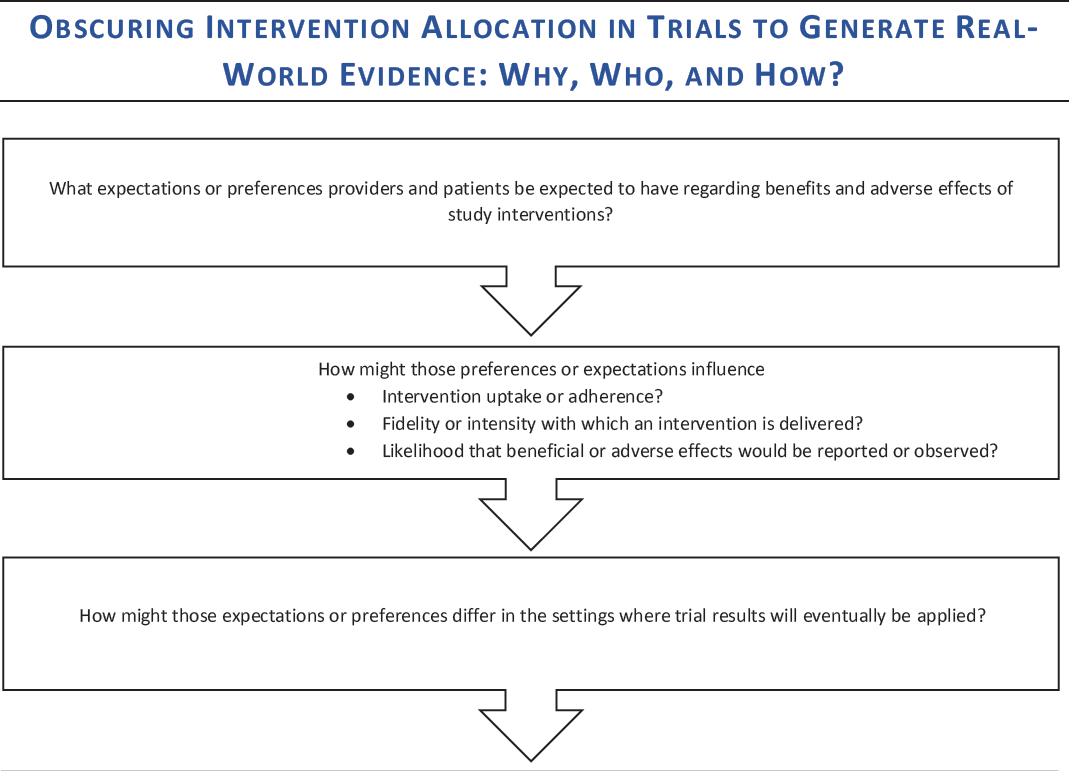
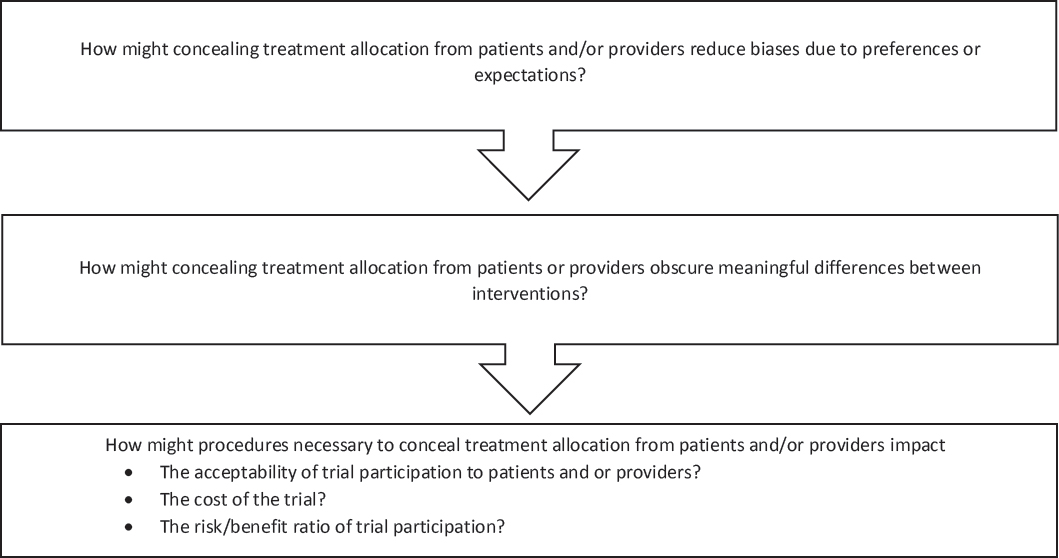
NOTE: This decision aid was drafted by some individual workshop participants based on the discussions of individual workshop participants at the first and second workshops in the real-world evidence series. The questions raised are those of the individual participants and do not necessarily represent the views of all workshop participants; the planning committee; or the National Academies of Sciences, Engineering, and Medicine, and the figure should not be construed as reflecting any group consensus.
SOURCE: Watanabe presentation, July 17, 2018.
choices in real-world evidence (RWE) study design. Participants at the third workshop reflected on these questions and offered feedback on the decision aid throughout the course of their discussions.
In addition to offering specific feedback on the decision aid (see Box 8-2), workshop participants also discussed the difficulty of answering these types of questions prior to conducting a study. Simon summarized that although the questions in the decision aid may be good questions to consider, making decisions about the risks and benefits of blinding in a study a priori is enormously difficult. Watanabe asked the participants if they thought that the answers to the questions on the decision aid could be quantified a priori—for example, if researchers could, before a study, quantify the risks and benefits of blinding and make a decision based on these numbers. Cathy Critchlow, vice president at the Center for Observational Research at Amgen Inc.; John Graham; and Vardeny all said that quantifying would be enormously difficult. Critchlow added that this sort of quantification could be risky because it could make the researchers “feel better about making a decision that may not be based on something real.” Richard Platt wondered if some of these a priori decisions could be based on empirical data on bias from previous research—for example, data on how aggressively vaccinated patients are treated versus non-vaccinated patients. Vardeny said that vaccinated patients are so different
from unvaccinated patients that it would be difficult to isolate the effect of any provider bias. Smith said an alternative approach could be statistically adjusting for the bias after the research was complete, which would still require a judgment, but may be conceptually easier than quantifying the bias a priori.
Simon suggested that there may be value, after a study is completed, in analyzing decisions made in the study to learn from any mistakes that may have been made. Smith concurred and said that “an autopsy of trials that do not go the way one expected can only be an incredibly valuable exercise,” and could be a best practice. Graham added that trial “autopsies” could be done not just for trials that gave a surprising result, but for all trials. He said that researchers could spend more time looking back at their decisions and the impact the decisions had on the results, and could include this information in their published results. Each study can give researchers insight into how to improve the next study, he said.
DISCUSSION
Context of the Decision
Critchlow echoed an earlier discussion (see Chapter 6) in which participants stressed the importance of considering the context of the decision when designing a study that uses real-world data (RWD). The decision about whether or not to blind participants in a study, she said, should consider what the study is meant to accomplish: Is it to answer a question about effectiveness, is it to inform treatment guidelines, is it to get approval for a new treatment, or is it to expand the label indications for an already approved treatment? Depending on the use of the study, blinding may be more or less appropriate. Researchers, she said, need to ask themselves: Is blinding helping us to answer the right question and make the right decision? Nancy Dreyer, chief scientific officer at IQVIA, agreed with this framing, and added a more specific question: What is the expected effect estimate, and how likely is it to be missed? For example, if a study that will use RWD is about a chronic disease for which the treatments might make a small but important difference, blinding might be necessary to get the needed effect size for the decision to be made, she said. Smith agreed that making a decision about blinding depends on the research question. For example, he said, if the research is seeking answers about whether patients are more likely to adhere to an injectable therapy or to an oral daily tablet, a blinded study cannot answer that question.
London concurred with the other speakers, saying that trial design decisions should be based on the specific uncertainties that a study is aimed at mitigating (see Box 8-3).
For example, if there are uncertainties about efficacy, blinding may be necessary, whereas if there are uncertainties about effectiveness in the real world, blinding would not be warranted. London explained that real-world effectiveness is a function of two variables: ensemble efficacy and utilization factors.
- The term “ensemble efficacy” acknowledges the fact that a drug is not efficacious on its own—the therapeutic effect depends on the proper dose and schedule, the right population, and co-interventions or diagnostic requirements.
- Utilization factors include patient and provider awareness and preferences, cost, adherence, clinical capacity, and tolerability. Even if an ensemble is efficacious, said London, the real-world effectiveness of the drug depends on the utilization factors. For example, a drug could be efficacious for reducing blood pressure, but if it is unaffordable, difficult to adhere to, or poorly tolerated, it will not be effective in the real world because patients will not use it.
Blinding may be more or less appropriate, depending on the state of knowledge about the ensemble efficacy and utilization factors, said London. If two interventions are being compared for real-world effectiveness, and it is known that Intervention 1 is more efficacious than Intervention 2, but the utilization factors are unknown, blinding would not be warranted (see Figure 8-3). The research question in this scenario, said London, is how these real-world factors such as patient preference will impact the effectiveness of an intervention. If patients and providers were blinded, the utilization factors would remain unknown and thus the real-world effectiveness would not be understood.
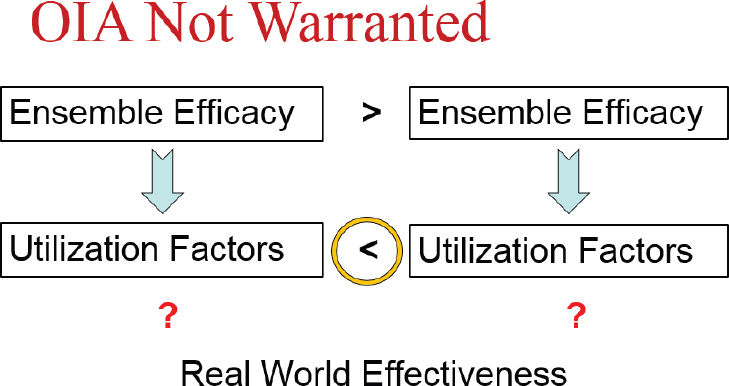
NOTE: OIA = obscuring intervention allocation.
SOURCES: London presentation, July 17, 2018; concepts from Kimmelman and London, 2015, and Moseley et al., 2002.
However, he said, a measure of real-world effectiveness may not always be sufficient for making policy or care decisions, compared to a measure of efficacy. London gave the example of arthroscopic surgery for osteoarthritis of the knee. This procedure, he said, was performed about 650,000 times annually at a cost of $3.25 billion. However, when a randomized controlled trial was performed to compare the procedure to a sham procedure, it was found to have no benefit (Moseley et al., 2002). The sham procedure, said London, was “largely theater” (manipulations, surface cuts, and bandages), but was necessary to obscure the intervention allocation. In this case, blinding was essential for a clear picture of intervention efficacy, and these efficacy data were necessary to determine the clinical merit of the procedure.
Risks of Blinding
When blinding is necessary to fully understand the effectiveness of an intervention, participants in the control group receive some sort of placebo drug or intervention, London said. This could range from a sugar pill, to a sham surgery that is “largely theater,” to invasive surgical procedures such as implanting cannulas in the brain to deliver saline in Parkinson’s trials, said London. There are risks to these procedures, and the benefits of a placebo are limited. The risks of these control procedures need to be disclosed to patients, he said. However, it is important to note that there
are also risks involved with the active procedure, and there may also be no benefit. For example, while there are minor risks involved with a sham knee surgery, the real knee surgery is far more invasive and risky. If the real knee surgery does not have any benefit, London said, the risks far outweigh the benefits. Both the risks from the placebo or sham procedure and the risks from receiving a potentially ineffective active intervention should be communicated to participants, London said. (See Chapter 7 for more discussion of patient safety in real-world studies.)
Another potential risk of blinding is that the participant population may be more homogeneous and more tightly controlled, which may make the results less generalizable, said Adrian Hernandez. The process of enrolling in a trial, and consenting to all of the processes necessary for blinding (e.g., complying with regular monitoring), may filter out some populations of participants.
Blinding can “obscure the truth,” said Simon. “We fundamentally distort the nature of the treatment” by making two active treatments seem identical and by ensuring that the patient and provider experiences with the treatments are identical. For example, he said, if two treatments have very different dosing schedules, this difference may have an important bearing on the acceptability and adherence to those treatments, he said. While there are obvious advantages of blinding (e.g., mitigating bias), it can also constrain opportunities to learn about how a treatment will work in the real world.
In discussions about whether or not to blind, said Dreyer, much attention is given to the risk of bias if a provider knows to which treatment group a patient belongs. However, she said, there are risks to the patient if a provider does not know which treatment he or she is receiving. For example, a provider may not know what adverse effects to monitor, or when a change in health status may indicate a significant problem. Dreyer said that while blinding can have benefits for the study, these benefits need to be weighed against the risks to the patient.
Cost Considerations
Researchers, said Dreyer, aim to get precise answers to research questions and then compare their answers to those that others get in order to confirm or refute the original hypothesis. However, Dreyer said that as a consumer and generator of data, researchers should be looking to see if two estimates are close, and certainly in the same direction, rather than expecting them to match exactly. Rather than designing a study to generate the most exact answer—which can increase costs significantly—she said, researchers should consider the big picture of the RWD’s intended use, and design studies accordingly. Dreyer said the economic value of different
study designs could be considered in the designs; if the answer can be found with an open-label, real-world study rather than from a more expensive randomized blinded trial—and it can be shown that bias is unlikely to explain the observed effect—it may be a better use of resources to choose the less expensive design, she said. Smith had a slightly different perspective. He said that while blinding can certainly increase the cost of a trial, an unblinded trial may “give you results that nobody believes,” which wastes time and resources.
Practical Considerations
For some interventions, such as vaccines, blinding is quite feasible and does not overly complicate the study, said Dreyer. However, for other interventions that involve treatments that are complex, sequential, or require monitoring and dose adjustment, it may be impractical or impossible to blind while keeping patient safety in mind. There are situations, said Smith, where blinding is simply not feasible. For example, some drugs have severe side effects, and to try to duplicate these side effects in the control group would be impossible or unethical. Blinding is a “tool that can be valuable in a narrow set of circumstances,” Dreyer said, but it should not necessarily be employed in all circumstances.
Hernandez noted that blinding could have the advantage of keeping participants in a trial. A participant who does not know if he or she is getting the active treatment may be more likely to continue care, he said.
Conducting a blinded study in a real-world setting may be difficult, said Rob Reynolds, vice president and Global Health, Epidemiology, Pfizer. Blinded RCTs are difficult enough, he said, but in a real-world environment with diverse health care sites and patient populations, the feasibility challenges are even greater. Dreyer joked that “a lot of things are feasible if you spend enough money.” She added that when doing blinded studies in the real world, researchers generally perform the studies at academic centers that are more able to carry out this type of research. However, this may result in a lack of generalizability because these centers are often especially high-quality facilities with a high quality of care and follow practices that may not be routinely conducted in community settings.
Patient Preferences
One question on the decision aid asks, “How might procedures necessary to conceal treatment allocation from patients and/or providers impact the acceptability of trial participation to patients and/or providers?” Smith said the best way to answer this question is to ask the patients directly.
Smith relayed an experience he had working with patients with a rare disease. He consistently heard that it would be impossible to conduct a placebo-controlled blinded trial in this population, but when meeting with a group of patients directly, the patients said if blinding “will give you the best data if we would be willing to go on a placebo for 6 months, we are happy to do so.” Smith said that assumptions about patients’ willingness to participate in blinded trials may not always be accurate. Critchlow added that in a context where there are fewer therapeutic options, patients might be more willing to take the risk of participating in a trial, whereas in a context with plenty of existing therapeutic options, the risk–benefit analysis of patients and providers is quite different.
On another patient-related topic, John Burch, an investor with the Mid-America Angels Capital Network, asked whether participants in a blinded trial are ever able to get information about which treatment they received. “If patients are entitled to their own information,” he said, “does that include patient-specific data from research?” Graham said that from an ethical perspective, research is likely progressing to a point where patients will be able to get information about their treatment in a trial.
Biases
Blinding, said Smith, is one mechanism to reduce the possibility of bias that would affect the trial results. There are multiple types of bias that can be associated with treatment assignment, some of which are discussed below. Smith said bias is often subconscious, and it can be difficult to predict the direction and magnitude of the bias. Different patients or providers within the same trial might have biases that affect the results in different directions, he said. For example, if aware of treatment assignment, one patient may believe that a novel drug will help, and therefore may overstate its benefit. Another patient may believe that the standard of care is fine and could be skeptical of a company’s development of a novel drug. The direction and magnitude of various biases may differ depending on the specific disease area, type of intervention, or patient population. Overall, Smith said, “It is very difficult to predict how bias is going to creep in and what direction and magnitude it is going to be.”
Patient Perception
Without blinding, there is potential for bias due to patient perception, said Critchlow. For example, patient perception may impact the likelihood of complying with a treatment regimen as intended. Dreyer said that bias due to patient perception can differ, depending on the subjectivity of the measurements. For example, if the outcome of interest is hemoglobin A1C
levels, patient perception is unlikely to have an effect. On the other hand, if the outcome of interest is pain level as reported by the patient, this could be influenced dramatically by perception, she said. Dreyer noted that for some outcomes, patient perception likely has no effect—“dead is dead.” Graham added that depending on the time line of the study, patient perception bias may wane over time. For example, if a patient gets a vaccine and feels ill 3 weeks later, does the patient’s initial idea of what treatment he received still affect his likelihood of seeking medical care? Graham said this is a very difficult area to have a black or white answer.
Ascertainment Bias
If the people who are assessing outcomes are not blinded, said Critchlow, it can result in ascertainment bias. The potential for ascertainment bias may depend on whether the endpoints are subjective or objective. For example, if the endpoint is a quantitative laboratory result, it is unlikely to be biased by the assessor knowing to which group the patient is assigned. Outcomes assessors, said Smith, can be blinded to mitigate ascertainment bias, but it must be recognized that “they only assess what has been given to them.” For example, a blinded pathologist can read a biopsy specimen, with no knowledge of the clinical course or treatment. However, blinding the assessor does not automatically remove the potential introduction of bias. For example, although the pathologist may be blinded, the decision by the patient’s provider to conduct a biopsy could have been influenced by their knowledge of treatment assignment. In this scenario, said Smith, “the bias has already been introduced before the outcome got to the outcomes assessor.”
Treatment Bias
When providers are not blinded during a trial (i.e., double blinded), said Smith, there is the worry that patients in one group are being treated differently than patients in the other group. For example, if a provider knows that a patient is on a placebo for suicide prevention versus an active treatment, the provider may be more likely to closely monitor the patient for signs of suicidality. The bias can also run in the other direction—if, for example, a provider knows that a patient is receiving an active but not well-understood treatment, the provider might monitor more closely for adverse events.
Channeling Bias
Channeling bias occurs later in the life cycle of a product when the adverse events associated with a product are well known, Jesse Berlin said during the second workshop. Patients who are at higher risk for these events are prescribed a different product; when these high-risk patients experience the event, the other product “starts to look like it’s increasing risk.” For example, non-steroidal anti-inflammatory drugs (NSAIDs) are associated with an increased risk of myocardial infarction (MI). Because of this, patients who are already at high risk of MI may be prescribed acetaminophen instead of NSAIDs, which results in a larger-than-expected group of acetaminophen patients experiencing MI.
















