
Endemic Social Diversity
Within Natural Kin Groups of a
Cooperative Bacterium
![]()
SUSANNE A. KRAEMER*†AND GREGORY J. VELICER*
The spatial structure of genetic diversity underlying social variation is a critical determinant of how cooperation and conflict evolve. Here we investigated whether natural social groups of the cooperative soil bacterium Myxococcus xanthus harbor internal genetic and phenotypic variation and thus the potential for social conflict between interacting cells. Ten M. xanthus fruiting bodies isolated from soil were surveyed for variation in multiple social phenotypes and genetic loci, and patterns of diversity within and across fruiting body groups were examined. Eight of the 10 fruiting bodies were found to be internally diverse, with four exhibiting significant variation in social swarming phenotypes and five harboring large variation in the number of spores produced by member clones in pure culture. However, genetic variation within fruiting bodies was much lower than across fruiting bodies, suggesting that migration across even spatially proximate groups is limited relative to mutational generation of persisting endemic diversity. Our results simultaneously highlight the potential for social conflict within Myxococcus social groups and the possibility of social coevolution among diverse related lineages that are clustered in space and cotransmitted across generations.
_____________
*Department of Biology, Indiana University, Bloomington, IN 47405. †To whom correspondence should be addressed. E-mail: suskraem@indiana.edu.
Social evolution research seeks to explain the origin, maintenance, and diversification of both cooperative and competitive social traits. This goal requires understanding the character of social environments that mediate selection on these traits. The distribution of behavioral and genetic diversity within and across groups of social animals has received much attention (Krebs and Davies, 1997; Oxley et al., 2010; Waddington et al., 2010). In contrast, relatively little is known about the structure of diversity among natural groups of social microbes (Fortunato et al., 2003b; Vos and Velicer, 2006, 2008a; Gilbert et al., 2009; Köhler et al., 2009; Wilder et al., 2009; Wollenberg and Ruby, 2009). However, detailed knowledge of group composition is necessary for understanding the roles of mutation, migration, lateral gene transfer, genetic drift, and various forms of selection in shaping the evolution of social microbes in natural habitats.
Microbes engage in a wide range of social behaviors, both cooperative and antagonistic, that affect the evolutionary fitness of others (Velicer, 2003; West et al., 2007a; Nadell et al., 2009). Some of the most biologically complex forms of prokaryotic cooperation occur in the myxobacteria (order Myxococcales, ?-proteobacteria), which are best known for social development of multicellular, spore-bearing fruiting bodies in response to starvation (Shimkets et al., 2006). In particular, the predatory soil bacterium Myxococcus xanthus has become a model organism for the study of microbial sociality, including cooperative motility (Wu and Kaiser, 1995), social predation (Berleman and Kirby, 2009) and fruiting body formation (Shimkets et al., 2006), and its population biology (Velicer and Vos, 2009).
M. xanthus cells swarm in a coordinated manner through soil habitats in cohesive groups using two genetically distinct motility systems, one of which is obligately social [type IV pili-driven “S-motility” (Hodgkin and Kaiser, 1977; Wu and Kaiser, 1995)] and one of which allows individual cell movement (“A-motility”) (Hodgkin, 1979; Sun et al., 2011). Swarms of M. xanthus in the soil kill and lyse prey cells of other micro-bial species with secreted antibiotics and lytic enzymes (Rosenberg and Varon, 1984). Upon starvation, swarming cells aggregate and develop into multicellular fruiting bodies (Shimkets et al., 2006). In these fruiting body aggregates a minority of cells convert to metabolically quiescent spores, whereas many other cells within the fruiting body lyse, possibly to the benefit of sporulating cells (Nariya and Inouye, 2008). The precise advantages of sporulation within fruiting bodies are unknown, although several hypotheses have been proposed, including enhanced dispersal, increased germination and/or growth rates in high-density groups, and protection from predation and/or environmental insults [summarized in greater detail in Velicer and Vos (2009)]. Here “fruiting body group” and
“group” generally refer to either all cells that compose a particular fruiting body or a set of laboratory strains isolated from the same fruiting body.
Although bacterial growth by binary fission in structured habitats inherently generates clonal cell pockets (Nadell et al., 2010), cell groups forming Myxococcus fruiting bodies are not expected to be entirely clonal owing to mutation. The M. xanthus genome is large [>9 Mb (Goldman et al., 2006)] and fruiting bodies are thought to be constructed by ?100,000 cells (Shimkets et al., 2006). If the mean M. xanthus mutation rate is roughly similar to that of Escherichia coli [?5.4 × 10–10 per base pair per generation in one estimate (Drake et al., 1998)], any given fruiting body should contain at least dozens of mutational variants, even if the entire fruiting body group originated from a single cell. Although most mutations are deleterious (Eyre-Walker and Keightley, 2007) and are lost by selection or genetic drift, a small minority will persist and rise to high frequency either because they confer a selective advantage or nonadaptively by hitchhiking (Maynard Smith, 1991) or genetic drift (Wright, 1931). Persisting mutants might be socially defective cheaters that increase owing to a frequency-dependent advantage within groups (Velicer et al., 2000; Fiegna and Velicer, 2003; Ross-Gillespie et al., 2007; Sandoz et al., 2007). Alternatively, such mutants may be socially proficient strains that outcompete dominant genotypes owing to increased intrinsic fitness (Buttery et al., 2009) or the ability to socially exploit majority genotypes (Strassmann et al., 2000; Fiegna and Velicer, 2005; Vos and Velicer, 2009). Assessing the degree to which such persisting mutants migrate across social groups—within which cells interact during motility, predation, and development—is critical for understanding social evolution in the myxobacteria.
If intergroup migration is low, within-group diversity should derive primarily from endemic mutation and be lower than diversity across groups. In this scenario, relatedness values for social loci among interacting variants may often be high, thus promoting the maintenance of cooperation by kin selection (Hamilton, 1964a; Sachs et al., 2004; Foster et al., 2006). Under low migration, cotransmission of within-group social diversity across generations will be high (Sachs and Bull, 2005; Wade, 2007), and lineages that repeatedly and preferentially interact may coevolve to reduce within-group conflict. Even if spatially proximate, distinct lineage groups among which migration is low may diversify via differential trajectories of adaptation and drift.
Previous work has indicated that some M. xanthus genotypes disperse far, despite the overall differentiation of meter-scale populations isolated by distance across large spatial scales [e.g., >2,000 km (Vos and Velicer, 2008a)]. Across smaller scales (<300 km), local meter-scale populations were not differentiated at the genetic loci examined, and dispersal
appears to be extensive (Vos and Velicer, 2008a). In another study, the spatial distribution of diverse multilocus genotypes among 78 centimeter-scale isolates did not appear to be significantly clustered, consistent with the possibility of extensive intergroup migration at this scale (Vos and Velicer, 2006). However, near the cellular (micrometer) scale, genetic variation in natural M. xanthus populations must be nonrandomly distributed due to the nature of bacterial colony growth by asexual binary fission in viscous environments (Nadell et al., 2010). Further work is required to better resolve patterns of genetic structure and degrees of dispersal and intergroup migration across a wide range of spatial scales.
A high level of phenotypic and genetic diversity has been documented among centimeter-scale M. xanthus isolates (Vos and Velicer, 2006, 2008b, 2009; Krug et al., 2008; Kraemer et al., 2010; Morgan et al., 2010), despite the fact that genetic diversity at this scale was found to be much lower than at only slightly larger sampling scales (Vos and Velicer, 2008a). For example, genetically similar centimeter-scale isolates were found to show extremely divergent competitive abilities during fruiting body development in forcibly mixed pairings (Vos and Velicer, 2009). However, the degree to which such diverse clones migrate across fruiting body–forming groups—either passively or by active motility—has remained unclear. In Myxococcus, cell–cell adhesion (Chang and Dworkin, 1994) and territorial kin discrimination (Vos and Velicer, 2009) may limit intergroup migration.
Using a new collection of natural isolates, here we have tested whether diverse social phenotypes coexist within the most discrete social unit of the Myxococcus life cycle, the fruiting body group. We then tested for group-level structure in genetic and phenotypic diversity to discriminate between scenarios of low vs. high migration among social groups residing in forest soils. Ten natural fruiting body groups were harvested from soil collected at three Indiana woodland locations separated by several kilometers. Fruiting bodies from a given kilometer-scale location originated from centimeter-scale (MC fruiting bodies; see Methods) or meter-scale (GH and KF fruiting bodies, see Methods) sites along sample transects. Forty-eight clones were independently isolated from each fruiting body and screened for diversity at several genetic loci and in several social phenotypes during group swarming and fruiting body development. Patterns of diversity within and across fruiting body groups were then analyzed. Detailed descriptions of the methods used can be found in Methods.
RESULTS
Variation in Swarming Phenotypes
Myxococcus group swarming on soft agar is a social trait that is driven by the type IV pili-based S-motility system in standard laboratory strains (Shi and Zusman, 1993). Natural fruiting body groups, each represented by 48 clones, varied significantly in their mean swarming rate on soft agar (Fig. 5.1A; Kruskal-Wallis test: P < 0.001). This result is consistent with previous work that documented extensive variation in soft-agar swarming among other natural isolates (Vos and Velicer, 2008b).
Seven fruiting bodies did not exhibit significant within-group variation in swarming rate according to Kruskal-Wallis tests (P >0.05), but two of these groups included stark variation in another visual phenotype (colony color in fruiting body GH5.1.9 and degree of cell–cell adhesion in MC3.3.5). Moreover, although swarming rate variation within GH5.1.9 was not quite significant according to the Kruskal-Wallis test (P = 0.08), comparison of the mean swarming rates of the two color types revealed an extremely significant difference (yellow vs. orange, Wilcoxon rank-sum test, P < 0.001).
Three fruiting bodies (KF3.2.8, KF4.3.9, and MC3.5.9) harbored clones exhibiting significant within-group variation in soft-agar swarming rate according to Kruskal-Wallis tests (Figs. 5.1 B–D and 5.2; P < 0.001 in all cases). Among KF3.2.8 clones, k-means clustering into two groups reveals one majority rate phenotype [mean cluster swarming rate 5.63 mm/d; 95% confidence interval (CI) = 0.23] and a faster minority type (mean cluster swarming rate = 6.32 mm/d; 95% CI = 0.3) (Fig. 5.1B). Post hoc testing revealed a significant difference between the swarming rates of those phenotype clusters (Wilcoxon rank-sum test on cluster means, P < 0.001), suggesting that they represent two distinct motility genotypes.
Cluster analysis of the other two fruiting bodies with significant variation among clones (KF4.3.9 and MC3.5.9; Fig. 5.1C and D) suggested the existence of three swarming rate phenotype classes in each group. The KF4.3.9 fruiting body is characterized by a fast majority type (mean cluster swarming rate 5.54 mm/d, 95% CI = 0.37) and two slower minority variants (Fig. 5.1C) (mean cluster swarming rates of 4.89 and 2.85 mm/d, respectively; 95% CI = 0.54 and 0.16, respectively). In contrast, the MC3.5.9 clones grouped into a majority phenotype with an intermediate swarming rate (mean cluster swarming rate 4.15, 95% CI = 0.47) as well as faster and slower minority types (Fig. 5.1D) (mean cluster swarming rates of 7.14 and 2.94 mm/d, respectively; 95% CI = 0.49 and 0.31, respectively). Post hoc testing revealed that the mean swarming rates among the clusters within both KF4.3.9 and MC3.5.9 differ significantly (Wilcoxon rank-sum tests of all possible within-fruiting-body combina-
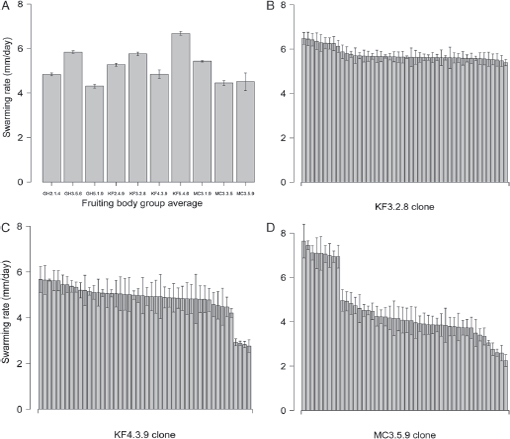
FIGURE 5.1 Swarming rates on soft agar. (A) Average swarming rates of all clones from each fruiting body group (n = 46-48 per fruiting body). (B-D) Average swarming rates for individual clones from fruiting bodies KF3.2.8, KF4.3.9, and MC3.5.9, respectively. Error bars represent 95% confidence intervals.
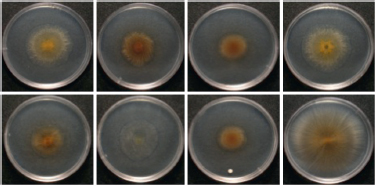
FIGURE 5.2 Swarming phenotypes of fruiting body MC3.5.9 isolates after 5 days of growth on soft agar plates. Upper: Left to right, c16, c19, c22, and c25; Lower: Left to right: c31, c35, c36, and c48.
tions of cluster means: P < 0.001 in all cases). In two cases, within-group variation in swarming rate was accompanied by variation in one or more additional phenotypes (colony color in KF4.3.9 and colony swarm pattern, color, opacity and degree of cell–cell adhesion in MC3.5.9; Fig. 5.2, Table 5.1).
Variation in Spore Production
Fruiting body groups, each represented by a subset of the 48 clones per fruiting body examined for motility, varied significantly in their mean levels of spore production (Fig. 5.3A; Kruskal-Wallis test: P < 0.001). This result is consistent with previous work that documented extensive variation in several developmental phenotypes among natural M. xanthus isolates (Fiegna and Velicer, 2005; Kadam and Velicer, 2006; Vos and Velicer, 2009; Kraemer et al., 2010).
Five of the 10 fruiting body groups examined were found to harbor significant within-group variation in spore production (Fig. 5.3B–D). In all five cases clones that sporulated poorly were in the minority. Spore production by the lowest sporulator within each of these five groups ranged from ≈10-fold below the level of the dominant phenotype cluster (MC3.3.5; Fig. 5.3C) to complete inability to produce viable spores (MC3.5.9; Fig. 5.3D). The isolates from fruiting bodies that harbored variation in spore production clustered into two distinct groups within each respective fruiting body by k-means cluster algorithms based on the criteria described in Methods. In two cases (GH5.1.9 and KF5.4.6), post hoc tests to compare cluster means were not possible because one cluster contained only a single clone. In the three remaining groups, the differ-
TABLE 5-1 Allelic and Phenotypic Variation Within and Across Fruiting Bodies
| Fruiting Body | Clone | Swarm | Spore | Colony | Gene | ||||||
| Rate | # | Color | pilA | 0128 | 0176 | 0396 | 0533 | 4405 | |||
| GH2.1.4 | 9 | y | 1 | 1 | 1 | 1 | 1 | 1 | |||
| 16 | y | ||||||||||
| 25 | y | ||||||||||
| 35 | y | ||||||||||
| 40 | y | 2 | |||||||||
| 48 | y | ||||||||||
| GH3.5.6 | 2 | y | 2 | 2 | 2 | 2 | 1 | 2 | |||
| 11 | y | ||||||||||
| 22 | y | 3 | |||||||||
| 36 | y | 2 | |||||||||
| 40 | y | ||||||||||
| GH5.1.9 | 17 | y | 1 | 3 | 3 | 1 | 2 | 2 | |||
| 20 | o | ||||||||||
| 27 | o | ||||||||||
| 37 | o | ||||||||||
| 47 | Low | y | |||||||||
| KF2.4.9 | 10 | y | 4 | 2 | 4 | 3 | 3 | 3 | |||
| 17 | y | ||||||||||
| 21 | y | ||||||||||
| 38 | y | ||||||||||
| 42 | y | ||||||||||
| KF3.2.8 | 1 | y | 5 | 4 | 5 | 4 | 4 | 4 | |||
| 11 | Fast | y | |||||||||
| 12 | Fast | y | |||||||||
| 16 | Fast | y | |||||||||
| 20 | y | ||||||||||
| 26 | Fast | y | |||||||||
| 28 | Fast | y | |||||||||
| 29 | Fast | y | ||||||||
| 30 | Fast | y | ||||||||
| 35 | y | |||||||||
| 37 | fast | y | ||||||||
| 39 | y | |||||||||
| 45 | y | |||||||||
| 48 | fast | y | ||||||||
| KF4.3.9 | 1 | Slow | Low | t | 1 | 2 | 6 | 1 | 5 | 5 |
| 2 | Slow | Low | t | |||||||
| 3 | y | |||||||||
| 19 | y | |||||||||
| 23 | Slow | t | ||||||||
| 28 | y | |||||||||
| 30 | Slow | Low | t | |||||||
| 37 | y | |||||||||
| 40 | Slow | y | 2 | |||||||
| 44 | y | 1 | ||||||||
| KF5.4.6 | 4 | Low | y | 6 | 1 | 7 | 5 | 6 | 6 | |
| 11 | y | |||||||||
| 28 | y | |||||||||
| 29 | y | |||||||||
| 36 | y | 1 | ||||||||
| MC3.1.9 | 3 | y | 7 | 5 | 2 | 1 | 1 | 5 | ||
| 16 | y | |||||||||
| 25 | y | |||||||||
| 28 | y | |||||||||
| 47 | y | |||||||||
| MC3.3.5 | 4 | Low | t | 2 | 6 | 7 | 6 | 1 | 2 | |
| 8 | Low | y | 2 | |||||||
| 9 | y | |||||||||
| 16 | y | 1 | ||||||||
| 21 | y | |||||||||
| Fruiting Body | Clone | Swarm | Spore | Colony | Gene | |||||
| Rate | # | Color | pilA | 0128 | 0176 | 0396 | 0533 | 4405 | ||
| MC3.3.5 | 34 | y | ||||||||
| 45 | y | |||||||||
| MC3.5.9 | 2 | Fast | y | 1 | 6 | 7 | 6 | 2 | 2 | |
| 5 | y | |||||||||
| 11 | Fast | y | ||||||||
| 13 | Fast | y | ||||||||
| 15 | Fast | y | ||||||||
| 18 | y | |||||||||
| 22 | Slow | Low | t | |||||||
| 23 | Fast | y | ||||||||
| 25 | y | |||||||||
| 27 | Fast | y | ||||||||
| 29 | Slow | Low | t | |||||||
| 31 | Slow | y | ||||||||
| 33 | y | |||||||||
| 36 | Slow | Low | t | |||||||
| 39 | Slow | Low | t | |||||||
| 41 | Fast | y | ||||||||
| 44 | y | |||||||||
| 46 | Fast | y | ||||||||
| 48 | Fast | y | ||||||||
Notes: Qualitative phenotype categories and allelic states at sequenced loci are shown for several clonal isolates from each fruiting body, including all clones that exhibited clear minority phenotypes for swarming and/or development. Distinct alleles at each locus are distinguished by a unique allele number (with numbers shown for only one representative of each allele). "Fast" and "Slow" designate individuals with minority swarming phenotypes in a given fruiting body, and "Low" designates individuals within low spore production clusters as identified by Jr-means cluster analysis (Methods). Colony color phenotypes are also indicated (y, yellow; o, orange; t, tan).
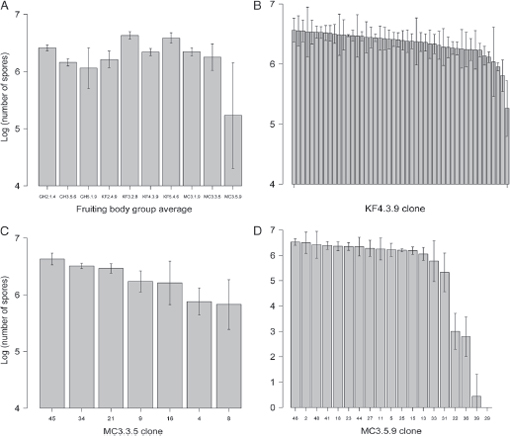
FIGURE 5.3 Spore production. (A) Average spore production of examined clones from each fruiting body group (n = 5-48 per fruiting body). (B-D) Average spore production by individual clones from fruiting bodies KF4.3.9, MC3.3.5, and MC3.5.9, respectively. Error bars represent 95% confidence intervals about means calculated from log10-transformed spore counts.
ences in spore production between the two clusters were either highly significant (KF4.3.9 and MC3.5.9; Wilcoxon rank-sum test, P < 0.001 in both cases) or marginally nonsignificant (MC3.3.5; Wilcoxon rank-sum test, P = 0.09).
Sporulation efficiency correlated significantly with swarming rate across all clones from all sampling locations (Spearman’s rank correlation p: S = 3,758, p = 0.56, P < 0.001).
Genetic Structure of Fruiting Body Groups
All clones assayed for spore production were screened for genotypic variation within ≈500 base pair windows of six loci that contain above-average levels of variation among the laboratory strain DK1622 (Kaiser, 1979) and two M. xanthus isolates from Tübingen, Germany [A23 and A47 (Vos and Velicer, 2006)]. Genetic variation within fruiting body groups was found to be extremely low relative to variation across fruiting bodies. Seven pilA alleles were found across all clones (Table 5.1), but only 5 of the 10 fruiting bodies harbored pilA polymorphisms, and no more than two pilA alleles were present in any fruiting body group. Each of the five other loci was highly polymorphic across fruiting bodies, with either six or seven alleles detected at each locus (Table 5.1). However, only one of these loci was found to vary among clones from the same fruiting body, in which instance a minority allele of Mxan_0533 was present in one clone of fruiting body MC3.3.5.
The vast majority of within-group phenotypic variation for swarming rate and spore production occurred between clones that are genetically identical at all (most cases) or most (a minority of cases) of the six loci examined (e.g., the swarming variants within fruiting body KF3.2.8 share the same alleles) (Table 5.1).
Phylogenetic Relationships
An unrooted maximum-likelihood tree and a Baysian inference tree were constructed with a sequence concatemer of the five loci other than pilA (Fig. 5.4). Both phylograms had similar topologies. Only one of the KF haplotypes (4.3.9) was found to group within the highly supported clade containing all of the GH and MC location haplotypes, with the KF5.4.6, KF2.4.9, and KF3.2.8 haplotypes branching more deeply. As reflected by this deep branching pattern, the KF3.2.8, KF2.4.9, and KF5.4.6 haplotypes were found to have only 0, 1, and 2 loci, respectively, that share an allele with one or more other fruiting body haplotypes (Table 5.1). KF3.2.8 is most similar to the laboratory strain DK1622 (Kaiser, 1979; Goldman et al., 2006).
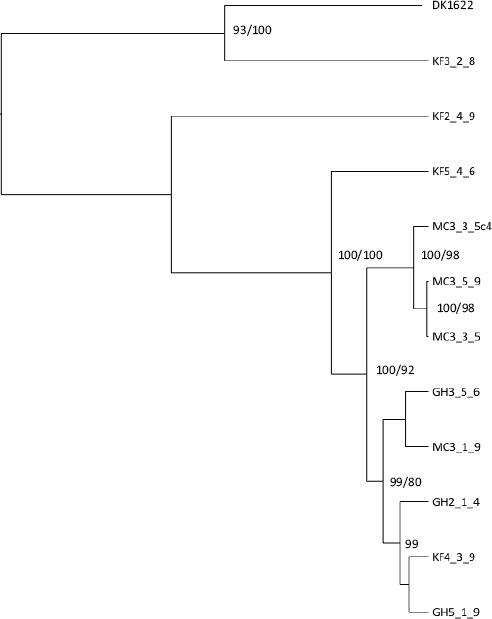
FIGURE 5.4 BI phylogram of 10 natural fruiting body groups based on a concatemer of the five loci (Mxan_0128, Mxan_0176, Mxan_0396, Mxan_0533, and Mxan_4405. BI and ML analyses produced similar topologies. Fruiting bodies harbored no internal variation at these loci with the exception of MC3.3.5, which contained a single minority variant MC3.3.5c4. Posterior probabilities >90 and bootstrap values >70 (based on 1,000 bootstrap replicates) are indicated at the nodes (posterior probabilities shown first).
Control for a Laboratory Origin of Minority Phenotypes
We tested whether the phenotypic variation demonstrated among isolated clones might have arisen during growth in the laboratory rather than in natural populations before soil collection. We examined four clones isolated from a fruiting body group (MC3.5.9) that exhibited a high degree of variation in both swarming rate and spore production. Specifically, we tested whether cultures from these four clones subjected to development and growth regimes similar to those experienced by the original fruiting body culture would generate an array of diverse phenotypes similar to that found among the 48 original MC3.5.9 clones. After cultures derived from these four clones had undergone development, heat, and sonication treatment and subsequent growth in CTT liquid, 48 clones were randomly selected from each culture after dilution plating and were screened for variation in swarming rate and fruiting body morphology. No significant variation was observed in either of these traits within any of the four clone sets, suggesting that the phenotypic variation documented here arose before fruiting body isolation.
DISCUSSION
The genetic and social diversity pervading natural Myxococcus populations is highly structured across local social groups within which cooperative—and likely antagonistic—interactions occur. This study and others have together shown that representatives of distinct but spatially proximate fruiting body groups vary starkly in social motility (Fig. 5.1A) (Vos and Velicer, 2008b) and several developmental phenotypes, including spore production (Fig. 5.3A), the rate of development, responsiveness to nutrient depletion in triggering development (Kraemer et al., 2010), and competitiveness in forced isolate pairings (Vos and Velicer, 2009). Here we have now also shown that pronounced social variation is present at high frequencies within many natural fruiting bodies—indeed within a majority of those fruiting bodies sampled here. Thus, diversity within fruiting bodies is high despite the fact that it was found to be much lower than diversity among fruiting bodies (Table 5.1 and Fig. 5.4). These results indicate that single clone isolates are likely to misrepresent the social phenotypes of other members of the groups from which they are isolated.
Possible Laboratory Effects
Control experiments strongly suggest that phenotypic variants isolated from the same fruiting body were already present at substantial frequencies in natural groups before sampling and did not arise in laboratory
cultures. However, our data do not necessarily reflect accurately the frequencies of these variants in the soil at the time of sampling owing to possible variation in growth rate under laboratory conditions. Natural isolates of bacteria might vary in their degree of “preadaptation” to laboratory conditions (Velicer and Lenski, 1999), and some degree of phenotypic variation may be specific to laboratory settings. Nonetheless, it is plausible that the large trait differences documented here reflect heritable variation that is also manifested during motility and/or development in natural habitats.
Phase Variation
The patterns of variation among fruiting body groupmates documented here are not explicable by a previously documented form of “phase variation” in M. xanthus. In phase variation, bacterial cells switch between discrete phenotypic states at much higher rates than would be generated by the genomewide average mutation rate (Laue and Gill, 1994, 1995; Beaumont et al., 2009). In M. xanthus laboratory strain DK1622, color phase variation occurs in which ≈1% of cells derived from a yellow colony grow into tan colonies, whereas ≈25% of cells from a tan colony grow into yellow colonies (Laue and Gill, 1994). Although it has been suggested that tan and yellow cells may have different functional roles during development (Laue and Gill, 1995), the genetic basis and population-level effects of M. xanthus phase variation remain poorly understood.
Most minority variants in swarming rate and sporulation among our fruiting body isolates did not exhibit minority color phenotypes (Table 5.1). The one exception is that all seven clones identified as having both unusually slow swarming and unusually low sporulation within their two respective fruiting body groups (KF4.3.9 and MC3.5.9) grew as tan colonies rather than as the majority yellow phenotype (Table 5.1). However, groupmates among these seven clones varied significantly in both swarming rate (Fig. 5.1C and D) and spore production (Fig. 5.3B and D), indicating that even these variants do not represent simple dual-state phase variation.
We screened 48 colonies derived from each of four clones isolated from the most internally diverse fruiting body, MC3.5.9. Three of the four parental clones (c6, c16, and c24) were yellow, and one was tan (c29). None of the 48 colonies derived from the tan clone were yellow, as would be expected for ≈25% of colonies under DK1622-like phase variation. Among these four clones only one instance of apparent phase variation was observed. In that case, the biphasic diversity observed differed dramatically both from the patterns of diversity documented among our original fruiting body isolates and from DK1622 phase
variation. Colonies derived from isolate MC3.5.9c6 showed two clearly distinguishable phenotypes of colony opacity during social swarming. Importantly, cultures derived from colonies of both opacity types form robust fruiting bodies and do not vary significantly in swarming rate. This limitation of variation among MC3.5.9c6 cells to colony opacity contrasts starkly with the variation observed among the original isolates from fruiting body MC3.5.9, which did not include similar variation in colony opacity but did include three distinct swarming-rate clusters and clones with severe developmental deficiencies. Colonies derived from the other three parental clones selected for the control experiments showed no variation for any visual phenotype. Thus, exhibition of phase variation is itself yet another phenotype that seems to vary among closely related groupmates within natural fruiting bodies.
Endemic Variation
Our results show that much of the detectable diversity within natural Myxococcus social groups derives from endemic mutation rather than intergroup migration. Here we consider migration to be the combination of dispersal to a new location and physical immigration into a new social group at that location. Five fruiting bodies, including the most phenotypically diverse one (MC3.5.9), contained only a single haplotype for all six loci sequenced here. Another four were polymorphic only at the pilA locus, with just two alleles present in each case. Only one fruiting body was polymorphic at more than one locus (MC3.3.5, polymorphic at pilA and Mxan_0533). In contrast, in almost all pairwise comparisons of fruiting body groups, the dominant six-locus haplotypes from the paired groups differed at most loci. The sole exceptional comparison is between MC3.3.5 and MC3.5.9, which share identical majority haplotypes. However, genetic variation seems to be structured even across these two fruiting bodies that were isolated at the centimeter scale because variants with similar phenotype profiles are represented by multiple clones within each fruiting body group but are absent from the other (Table 5.1).
In contrast to patterns revealing endemic variation, the occurrence of some alleles that are shared by clones from multiple fruiting bodies isolated from different locations is consistent with some degree of recombination across groups and populations, possibly mediated by phage transduction (Martin et al., 1978). Such patterns are not unexpected in light of previous evidence for horizontal gene transfer in Myxococcus populations (Vos and Velicer, 2006, 2008a; Vos and Didelot, 2009).
Migration
Our data suggest that the rate at which Myxococcus social variants arise by mutation and subsequently increase to detectable frequencies within their natal groups is high relative to the rate at which variants migrate into “foreign” groups and subsequently persist. The among-group migration rate will greatly affect the relative importance of within-vs. among-group selection (Wade, 1985) in determining the fate of new mutations in social genes. Low migration will promote spatiotemporal clustering of genetically similar lineages [i.e., high relatedness within groups (Foster et al., 2006; Gilbert et al., 2007, 2009)], high cotransmission of social diversity across generations (Wade, 2007), and the among-group component of selection in multilevel selection models of social evolution (Wade, 1985).
Biological traits that affect migration rate are thus likely to influence how cooperation is maintained in a metapopulation and the evolutionary forces causing socially proficient genotypes to diversify. In Myxococcus, most cells are highly cohesive owing to the production of cell-surface adhesins, which should hinder emigration away from natal kin groups. Moreover, kin discrimination mechanisms that hinder immigration (Travisano and Velicer, 2004) by members of neighboring groups appear to be pervasive in natural M. xanthus populations. This inference derives from experiments in which neighboring swarms of genetically very similar centimeter-scale isolates failed to merge on agar plates for most pairings (Vos and Velicer, 2009) (Fig. 5.5). Cooperation benefits buttressed by low migration may thus contribute to selection for cell–cell adhesion and territorial kin discrimination.
Limitation of Socially Defective Cheaters
Cheater strains with social defects in clonal groups that can exploit cooperative genotypes in mixed groups during Myxococcus development readily appear by mutation in laboratory populations (Velicer et al., 2000). When rare, these cheaters have a within-group advantage over cooperators but lose that advantage and impose cheating load (i.e., reduce group productivity) (Velicer, 2003) when they reach high frequencies (Velicer et al., 2000; Fiegna and Velicer, 2003). Many isolates described here exhibit low spore production (e.g., MC3.5.9c29; Fig. 5.3D) and/or slow swarming (e.g., KF4.3.9c1; Fig. 5.1C). These clones may represent cheaters of natural origin that defect from “fair” production levels for a social compound required for development or social motility but exploit others who produce more of that compound. Alternatively, socially deficient strains may be present owing to genetic drift or selection on some trait other than the socially defective one.
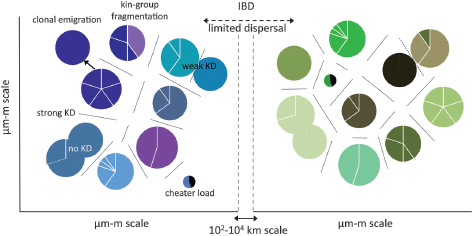
FIGURE 5.5 Simple hypothetical model of natural Myxococcus population biology. Circles represent social groups within which individuals directly interact. Sectors represent genetically distinct within-group variants. Distinctly shaded circles represent among-group genetic differentiation, with lines separating kin discrimination (KD) units (Vos and Velicer, 2009). Overlapping circles represent lack of KD between highly similar, but nonetheless genetically distinct, social groups. Multishaded circles represent kin-group fragmentation (see text). Small circles with a black sector represent cheater-infected groups burdened by cheater load (Velicer et al., 2000; Fiegna and Velicer, 2003; Velicer and Vos, 2009). Left vs. Right panels represent population differentiation across large spatial scales due to isolation by distance (IBD) (Vos and Velicer, 2008a). The arrow at Left represents establishment of a new clonal group by clonal emigration.
Only mutations creating socially defective cheaters that have an advantage within a group across an organism’s entire life cycle will increase substantially within groups. The mutation rate to cheaters that have such a net within-group advantage (at least when rare) remains unknown. Pleiotropy may limit this mutation rate (Foster et al., 2004; Travisano and Velicer, 2004). Cheater mutations that are net beneficial within groups may nonetheless be net deleterious across groups in a larger metapopulation owing to among-group selection mediated by cheater load (Velicer et al., 2000; Fiegna and Velicer, 2003; Gilbert et al., 2007; Velicer and Vos, 2009) and promoted by limited migration (Fig. 5.5).
The mutation rate to socially defective cheating alleles that are net beneficial within groups might be lower than the rate at which such alleles are lost from a metapopulation owing to their net deleterious effect at the among-group level. In this scenario, the overall frequency of socially defective cheaters in a metapopulation should be determined
by the relative magnitude of that mutation rate and the strength of among-group selection against the spectrum of cheaters that arise by mutation (Crow and Kimura, 1970; Van Dyken et al., 2011). Alternatively, if the combination of mutation rate to socially defective cheaters and intergroup cheater migration is sufficiently high relative to the rate of cheater loss via among-group selection, all social groups in a metapopulation could become infected by cheaters. Four of the 10 Myxococcus fruiting body groups examined here did not harbor variation in spore production or social swarming at frequencies above our detection limits, suggesting that these groups did not harbor cheaters at the time they were sampled. This result is consistent with (but not demonstrative of) the possibility that socially defective cheater frequencies in natural Myxococcus populations are largely determined by “kin selection–mutation balance” (Van Dyken et al., 2011), with kin selection in this scenario being mediated by selection among spatially structured kin groups.
Coevolution
High cotransmission of within-group diversity across generations aligns the evolutionary interests of clustered lineages (Sachs and Bull, 2005; Wade, 2007). Under low migration, diverse lineages that repeatedly and preferentially interact may coevolve to reduce conflict (Bouma and Lenski, 1988; Stewart et al., 2005; Weeks et al., 2007) and chimeric load (i.e., reduced group productivity caused by within-group diversity). Cheating load is one form of chimeric load. If coevolution within cheater-infected groups proceeds rapidly relative to the rate of cheater loss by among-group selection, immunity to socially defective cheaters and policing behaviors that suppress them may evolve, as has occurred in experimental populations (Fiegna et al., 2006; Zhang et al., 2009; Manhes and Velicer, 2011). Indeed, cheaters themselves may reevolve proficiency at cooperation by novel genetic routes (Manhes and Velicer, 2011) and thereby perhaps reach new cooperation fitness peaks (Wright, 1932) not accessible to noncheaters.
Clusters of socially cotransmitted lineages may also coevolve to reduce chimeric load generated by behavioral incongruities among interacting strains that are each socially proficient in clonal groups (Castillo et al., 2005; Fiegna and Velicer, 2005; Vos and Velicer, 2009). Cotransmitted lineages might even coevolve to perform distinct mutually beneficial functions that raise cooperative group productivity beyond that achievable by clonal groups. Unique trajectories of coevolution among clusters of cotransmitted lineages may promote diversification across kin groups.
Regeneration of Clonality
New clonal groups can be established by clonal emigration from an internally diverse group (Fig. 5.5) (Travisano and Velicer, 2004) or by local selective sweeps of adaptive mutants that purge variation from an existing kin group (Cohan, 2001). Alternatively, new kin discrimination alleles might arise by mutations that generate biological barriers to migration across clonal cell patches (Nadell et al., 2010) and thereby fragment a preexisting group into multiple kin discrimination units (Fig. 5.5). The rate at which clonal groups of cooperative genotypes are freshly established (or reestablished) is unknown but is an important parameter for understanding cheater–cooperator population dynamics.
What Maintains Diversity Within and Between Groups?
Natural variation in M. xanthus social traits documented here and previously (Krug et al., 2008; Vos and Velicer, 2008b; Kraemer et al., 2010; Morgan et al., 2010) may be nonadaptive and may have reached detectable frequencies by genetic drift or hitchhiking (Maynard Smith, 1991) or might reflect pleiotropic byproducts of evolutionary adaptation at some alternative trait. For example, even variation that causes large fitness differences during laboratory developmental competition experiments (Strassmann et al., 2000; Fiegna and Velicer, 2005; Vos and Velicer, 2009; Saxer et al., 2010) need not have been shaped by selection for within-group competitiveness during development. Indeed, the strongest developmental cheaters yet identified in M. xanthus originated in a selective regime in which evolving populations never underwent development (Velicer et al., 1998, 2000). Alternatively, selective sweeps may be driving some observed variants to fixation. Finally, Myxococcus social diversity may be maintained by various forms of balancing selection.
Within kin groups, frequency-dependent selection might maintain both cooperators and socially defective cheaters (Velicer et al., 2000) or multiple genotypes that mutually benefit one another owing to differential expression of cooperative traits (Manhes and Velicer, 2011). Within-group diversity might also be promoted by specialized performance among genotypes across variable environmental conditions [e.g., surface conditions (Shi and Zusman, 1993; Hillesland and Velicer, 2005; Vos and Velicer, 2008b), prey composition (Morgan et al., 2010), etc.]. Across kin groups, balancing selection might take the form of kin-group specialization to different microhabitats or nontransitive fitness relationships (Kerr et al., 2002) during competitive interactions, such as the production of anticompetitor compounds (Riley and Gordon, 1999) by adjacent kin groups.
The extensive diversity within natural Myxococcus social groups documented here suggests that within-group conflict is likely to play a major role in myxobacterial social evolution. Migration among kin groups seems to be low relative to the rate at which persisting variants arise by mutation and coevolution among socially cotransmitted Myxococcus lineages is likely to occur. The relative roles that the fundamental forces of evolution—mutation, distinct forms of selection, migration, genetic drift, and recombination—play in shaping natural social variation in the myxobacteria remain to be quantified. Doing so will require estimation of mutation rates, identification of loci and alleles responsible for observed social variation, screening for population genetic signatures of distinct evolutionary forces, and characterization of fitness relationships among social interactants under conditions relevant to natural habitats.
METHODS
Sample Collection and Strain Isolation
Soil samples were collected in a spatially nested design at three undisturbed woodland locations near Bloomington, Indiana [Old Meyers Road (GH) and Indiana University teaching and research preserves at Kent Farm (KF) and Moores Creek (MC)]. At each location, five sample sites were established at 10-m intervals along a line. At each of these meter-scale sample sites, five soil samples were collected at 2-cm intervals along the line. Samples were collected as described previously (Vos and Velicer, 2006) with a sterile 2-mL syringe from which the tip had been removed. Syringes were sealed with parafilm immediately after sampling to avoid cross contamination. After sampling, syringes were stored overnight at room temperature.
The day after sampling, ≈2 mm were removed from the ends of each soil core with a sterile scalpel, and the remaining core was crumbled onto selective agar medium [CTT medium with 1.5% agar (Hodgkin and Kaiser, 1977) containing the antibiotics and antifungals vancomycin (10 mg/L), nystatin (1,000 units/L), cyclohexamide (50 mg/L), and crystal violet (10 mg/L)]. Plates were incubated at 32 °C, 90% rH. After 2 weeks, plates were examined for the presence of fruiting bodies on soil particles. Ten spatially separated and individually discrete fruiting bodies were picked with a sterile toothpick from each plate. Each fruiting body was placed in a separate microcentrifuge tube containing 0.5 mL ddH2O and heated at 50 °C for 120 minutes to kill nonspore cells. Samples were sonicated twice for 10 seconds to disperse spores and then transferred to CTT growth medium. Early samples (GH2.1.4, GH3.5.6, KF5.4.6, MC3.3.5) were transferred into CTT liquid and grown at 32 °C, 300 rpm, as were all liquid
cultures described below. Cultures were grown until exponential phase (1–3 days) and then frozen with 20% glycerol at -80 °C (as were all frozen samples). Assuming a generation time of 4 hours [most likely a conservative underestimate (Velicer et al., 1998)], these cultures underwent no more than 18 generations of growth from the original group of fruiting body spores harvested directly from soil until frozen storage.
However, because of several instances in which contaminants that survived the heat and sonication treatments outgrew Myxococcus cells in liquid culture, subsequent samples (GH5.1.9, KF2.4.9, KF3.2.8, KF4.3.9, MC3.1.9, MC3.5.9) were transferred onto CTT hard (1.5%) agar after sonication and incubated at 32 °C, 90% rH (as were all agarplate cultures described below). Plates were screened after 3 to 5 days for growth of Myxococcus cells, which grow into swarming colonies that are easily distinguished from contaminant colonies. When no contaminants were present, the entire Myxococcus population was harvested with a sterile scalpel and transferred into CTT liquid. If contaminant colonies were present, as much of the Myxococcus population as possible was harvested without touching contaminant colonies. Liquid cultures were incubated overnight and frozen. Cultures that underwent growth on agar plates likely underwent no more than 36 generations of growth from the original group of fruiting body spores harvested directly from soil until frozen storage.
Thawed samples (10 μL) from each of 10 frozen stocks derived from fruiting bodies isolated from the three sampling locations were diluted with CTT liquid into CTT soft (0.5%) agar (at 40 °C) at several dilution factors. Forty-eight spatially distinct colonies from each fruiting body culture were inoculated into separate flasks of CTT liquid, grown to high density, and frozen.
Fruiting body names reflect the sample location (GH, KF, or MC) and position (numbers). The first and second numbers in each name identify the meter- and centimeter-scale positions from which the respective soil sample was taken and the third number identifies the particular fruiting body taken from a given soil sample. Fruiting bodies from the GH and KF locations examined here were isolated from soil particles separated by meters in the sample plot, whereas the soil particles from which the MC fruiting bodies were isolated were from the same centimeter-scale plot because soil from other locations along the meter-scale transect did not yield fruiting bodies.
Swarming Motility Assays
Cells from all 48 clones representing each isolated fruiting body were inoculated from frozen stocks into 8 mL CTT liquid and incubated for 3 or 4 days. Cultures that reached exponential growth phase prior to oth-
ers were diluted to avoid entry into stationary phase. The day prior to the swarming assay, cultures were diluted to 3 × 107 cells/mL. CTT soft (0.5%) agar plates were poured on the same day (25 mL in 9-cm-diameter petri dishes) and allowed to solidify uncovered for 15–20 min in a sterile laminar-flow hood before being covered and stored overnight at room temperature.
To initiate the swarming assays, 5 mL of each exponential-phase culture were centrifuged at 4,500g for 15 minutes and then resuspended with CTT liquid to 5 × 109 cells/mL. Ten microliters of each resuspended culture was then placed at the center of an agar plate and subsequently plates were incubated for 5 days. Swarm perimeters were marked after 1 and 5 days of incubation, and the distance swarmed between those time points for each replicate was measured as the average distance along four perpendicular vectors at a random orientation. More vectors were used for irregularly shaped swarms. All experiments (also those below) were performed in at least three temporarily independent replicate blocks.
Sporulation Assays
Five clones each were assayed for spore production from fruiting bodies that did not exhibit variation in swarming rate or other motility phenotypes. For fruiting bodies that did show variation, five clones of the majority swarming phenotype and minority-phenotype clones were assayed for spore production. For one fruiting body (KF4.3.9), all clones were included in the sporulation assay. Frozen samples were inoculated into CTT liquid, and resulting cultures were grown to visible turbidity but prevented from entering stationary phase by dilution if necessary. To initiate development, culture samples were centrifuged and resuspended to ≈5 × 109 cells/mL in TPM liquid (a buffered medium with no added carbon source) (Kroos et al., 1986). Ten microliters of each culture was spotted onto TPM hard (1.5%) agar plates and incubated. After 3 days, spores were harvested from the agar surface with a sterile scalpel, transferred into 1 mL ddH2O and heated for 2 hours at 50 °C. After heat treatment spores were sonicated twice for 10 seconds and then diluted into CTT soft (0.5%) agar previously cooled to 40 °C. Plates were incubated 1 week, after which colonies were counted.
Control for Laboratory Origin of Minority Phenotypes
We tested the hypothesis that minority variants observed in our motility and sporulation assays might have originated by mutation during culture growth in the lab rather than in natural populations prior to soil collection. To do so, we tested for phenotypic variation within cultures
derived from four randomly chosen clones isolated from fruiting body MC3.5.9, which exhibited a high degree of within-group variation in both motility and sporulation phenotypes. Cultures of the MC3.5.9 clones were subjected to growth in liquid medium (3–4 days), one cycle of development on TPM agar followed by heat and sonication treatments and subsequent growth again in CTT liquid prior to being diluted into CTT soft agar to allow isolation of clones for phenotypic analysis. Forty-eight clones from each initially clonal culture were isolated at random and examined for variation in swarming motility rate and phenotype as well as variation in fruiting body phenotypes after 10 μL of culture (5 × 109 cells/mL) were spotted onto CF hard (1.5%) agar (Hagen et al., 1978) and incubated for 5 days. Average swarming rates and photographs of fruiting bodies for all control clones are available upon request.
Statistics
All statistical analyses were performed with R software (R Development Core Team, 2009). Sporulation data were log10-transformed before analysis. Clones in populations harboring significant levels of variation were partitioned into phenotype clusters using k-means cluster algorithms. Specifically, optimal cluster values were selected by minimizing the within-cluster sum of squares (Everitt and Hothorn, 2009). If three clusters were present, nonindependent post hoc tests were performed with Bonferroni corrections.
DNA Sequencing and Phylogenetic Analysis
Five randomly selected clones representing the majority phenotype within each fruiting body were chosen for comparative DNA sequence analysis, as were (nonrandomly selected) clones that exhibited clearly distinct minority phenotypes in motility, sporulation, colony color, or degree of cell-cell adhesion. Approximately 500-bp fragments of six loci were sequenced for selected clones: pilA and five loci [loci Mxan_0128, Mxan_0176, Mxan_0533, Mxan_0396, and Mxan_4405 (Goldman et al., 2006)] identified as being highly variable among the M. xanthus strains DK1622 (Kaiser, 1979; Goldman et al., 2006), A23 and A47 (Vos and Velicer, 2006) based on unpublished whole-genome sequence comparisons. Primer sequences and details of PCR and sequencing reactions are available upon request. All sequences were aligned with CodonCode Aligner Version 3.7.1 (CodonCode, Deadham, MA) and adjusted manually. Sequences are deposited at GenBank under the accession numbers JF819182–JF819591 and JF741968–JF742049.
Phylogenentic analysis was based on a 2,270-base concatemer of all loci sequences except pilA, which is more polymorphic than the other loci. Independent phylogenetic analyses were performed using maximum likelihood (ML) and Bayesian inference (BI) with Kimura-2 parameters of base substitution. We determined the ML phylogram using Mega Version 4.0 (Tamura et al., 2007) and assessed its support using 1,000 bootstrap replicates.
BI analyses were performed in BEAST version 1.6.1 (Drummond and Rambaut, 2007). Two MCMC runs with trees sampled every 1,000 generations were performed for 10 million generations and subsequently combined. Convergence was assured by visual inspection of parameter sample plots in Tracer version 1.4 (Drummond and Rambaut, 2007) and the first 10% of the analysis was discarded as burn-in. Bootstrap values >70% and posterior probabilities >95 were counted as high clade support.
ACKNOWLEDGMENTS
We thank H. Peitz for sharing genomic data; T. Platt for helpful suggestions regarding the control experiments; and E. Hanschen and M. Toups for helpful suggestions regarding statistical and phylogenetic analyses. This study was supported by National Institutes of Health Grant GM079690 (to G.J.V.). The sequences reported in this chapter are deposited in the GenBank database (Accession Nos. JF819182–JF819591 and JF741968–JF742049).
This page intentionally left blank.


























