
Vega-Rua, A., K. Zouache, R. Girod, A. B. Failloux, and R. Lourenco-de-Oliveira. 2014. High level of vector competence of Aedes aegypti and Aedes albopictus from ten American countries as a crucial factor in the spread of chikungunya virus. Journal of Virology 88(11):6294-6306.
Villar, L., G. H. Dayan, J. L. Arredondo-Garcia, D. M. Rivera, R. Cunha, C. Deseda, H. Reynales, M. S. Costa, J. O. Morales-Ramirez, G. Carrasquilla, L. C. Rey, R. Dietze, K. Luz, E. Rivas, M. C. Miranda Montoya, M. Cortes Supelano, B. Zambrano, E. Langevin, M. Boaz, N. Tornieporth, M. Saville, F. Noriega, and C. Y. D. Study Group. 2015. Efficacy of a tetravalent dengue vaccine in children in Latin America. New England Journal of Medicine 372(2):113-23.
Yendell, S. J., M. Fischer, and J. E. Staples, 2015. Colorado tick fever in the United States, 2002-2012. Vector Borne Zoonotic Diseases 15:311-316.
A10
ARBOVIRUS EVOLUTION, VECTOR COMPETENCE, AND VIRULENCE MODELS—CHANGING PATTERNS OF INFECTION
Corey W. Hecksel and Rebecca Rico-Hesse1
Abstract
Viruses that are transmitted by arthropods, especially mosquitoes, have emerged very recently as major causes of public health concern: dengue and chikungunya viruses are transmitted by some of the most common mosquito vectors that cohabit with and bite humans, and new virus variants are being transmitted at increased rates throughout the world. Not only are these viruses spreading, along with humans that travel and infect other mosquitoes, but the most virulent variants are being naturally selected for and causing increased disease severity. Here we summarize the approaches used to measure these evolving arbovirus characteristics, what we might expect in the near future, and what we are doing to try to understand the mechanisms of their evolution and transmission, in order to design effective control measures and provide accurate input for mathematical models of disease dynamics.
Introduction
In the context of this workshop, it is important to discuss how mosquito-viruses have been evolving, so that we might find some patterns to guide our expectations of any type of emergence. Here we discuss what is currently known about two of the most important arthropod-borne viruses (arboviruses), dengue
___________________
1 Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, Texas, USA.
(DENV) and chikungunya (CHIKV), which are continuing to spread globally, and being introduced to new populations (both mosquitoes and humans) that are susceptible and are clinically inexperienced with the diseases these viruses cause. Although these viruses belong to different virus genera (Flavivirus and Alphavirus, respectively) and contain different genes and have different structures, they are both transmitted by bite, by the same two mosquito species, Aedes aegypti and Aedes albopictus (females only, as they require blood for egg development), forming mosquito–human–mosquito cycles of virus amplification and transmission. Both viruses contain single-stranded RNA genomes that are normally prone to higher rates of mutation than DNA viruses; however, these viruses evolve at lower rates than other RNA-containing viruses, presumably because they are constrained in maintaining their ability to replicate in humans and mosquitoes, with differing host cell receptors, cell biology, and mechanisms of immunity.
These two arboviruses produce large numbers of virus particles in the human host’s blood (viremia) after infection by mosquito bite (via saliva) and are therefore infectious to other biting vectors, such as mosquitoes, ticks, and flies. However, these two viruses are capable of replicating and spreading throughout the bodies and salivary glands of only the two above-mentioned Aedes mosquitoes, without causing mortality; thus, these mosquitoes are serving as exponential biological amplifiers for the viruses. Therefore, when we study the evolution of these viruses, we must evaluate how they replicate, disseminate, and are transmitted via saliva by mosquitoes; this is referred to as vector competence. In contrast, for human hosts, we need to determine rates of infection and replication, or pathogenesis and virulence in individuals with differing immune status, genetic predispositions to viral infections, and possibly other underlying diseases. Thus, for detecting differences between evolving viruses in humans, we require systems in which to measure virulence, or the effects of viral replication and the damage this causes in different human cells (tropism), leading to overall pathogenesis and differing degrees of disease; these factors would also have an impact on how often the human viremic host could infect other vectors. And, unfortunately for us, no other animal responds to DENV or CHIKV like humans, so we have no straightforward models in which to study replication, immunity, and clinical presentations.
To test these principles of arbovirus evolution and the ultimate effects on disease emergence, we need to measure evolutionary pressures and effects in all three organisms (virus, mosquito, and humans), requiring us to rely on very diverse assay systems: for the viruses, we study their genetic changes via nucleotide sequencing, by generating phylogenetic trees, and by generating structures, with reflected protein changes that might change tropism or escape immunity; for humans, we study epidemiological links to specific virus antigenic (serotypes) or genetic variants (genotypes), how the viruses behave in primary human cell culture, or in new, complex animal models of disease and virulence; and for mosquitoes, we can work with field-collected or colonized mosquitoes, which are
bred in the laboratory under controlled environments. Because of the complexity of these systems, it is important to note that the outcomes of these measurements and comparisons should always be done in systems that mimic the natural cycles of virus transmission; otherwise, these studies could lead to incorrect conclusions. These studies are necessary to determine if vaccination and treatment strategies, for which there are none currently licensed, can be directed at the most virulent virus variants that cause the majority of the disease outbreaks. This information can also help to strategize disease control approaches, which are even more critical when dealing with mosquito vectors that cannot be eradicated. The importance of these studies is also highlighted by the fact that these data are needed to derive any of the mathematical models of disease dynamics also discussed in this workshop, which depend on accuracy of input for realistic prediction outcomes.
Dengue Viruses
Disease Characteristics and Viral Genetic Variability
Currently, dengue viruses are the most prevalent of all the arboviruses, causing an estimated 400 million human infections per year, in over 100 countries worldwide; however, these infections produce clinical disease, dengue fever (DF), in only around 10–15 percent of those infected (attack rate) (for reviews see Bhatt et al., 2013; Guzman et al., 2010). The most recent incursion of new virus variants into the Americas, during the 1980s, caused the first massive epidemics of dengue hemorrhagic fever (DHF), which is the most severe form of the disease, and can be fatal in 5–20 percent of patients. Because dengue viruses differ by around 25 percent in their exterior proteins, we classify them into four different antigenic groups or serotypes; this also means that immunity to one serotype does not protect against infection by another serotype. In fact, this insufficient “secondary” immune response increases the possibility of getting more severe disease, or DHF. So, in addition of having to measure direct destruction of cells by the virus, we also have to measure the effects of immunologic responses gone wrong; these events were first described as antibody-dependent enhancement of disease, but we now know that other immune responses, such as cytokines/chemokines secreted during an inflammatory response, lead to platelet and endothelial cell activation that cause vascular leakage and hemorrhaging (for a review, see Rothman, 2011). In addition to having four different serotypes, each of these serotype viruses show variability in their genomes’ sequences, and can be classified into genotypes; there are three to five genotypes within each serotype, and these have specific geographic distributions, epidemiology, virulence, and/or transmission abilities associated with each of them (for review see Rico-Hesse, 2003). Therefore, it is very important to measure these evolving differences between strains or variants, because these characteristics can be closely associated with potential for spread and human clinical disease presentations.
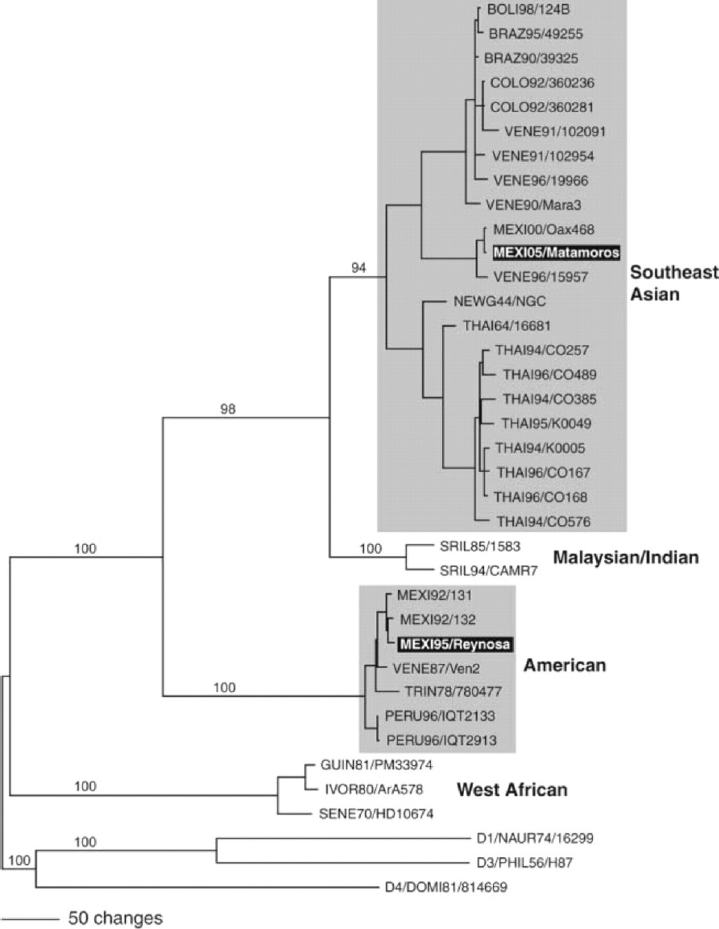
Phylogenetic trees DENV contain approximately 11,000 nucleotides of RNA, and produce 8 different proteins, in the order 5′-capsid-membrane-envelope-non-structural proteins 1 through 5-3′; in addition, they have around 100 nucleotides at the beginning (5′) of their genome, and 400 nucleotides at the end (3′), that do not code for any proteins (known as untranslated region; UTR). We showed some time ago that nucleotide sequences from certain areas of the DENV genome could be used to construct evolutionary trees that could help us separate out variants from different regions of the world and demonstrate their times and routes of spread (Rico-Hesse, 1990; Rico-Hesse et al., 1997). Since then, numerous studies have been published comparing different areas of the viral genome or complete genome sequences for various DENV strains, and are continuously being updated along with epidemiologic characteristics. To this end, we will focus on the interpretation of one type of phylogenetic tree, for DENV of serotype 2, to demonstrate the first linkages of genotypes with certain epidemiologic and virulence characteristics.
In Figure A10-1, a phylogenetic tree constructed with the nucleotide sequences from the envelope gene of numerous DENV-2 strains, and representatives of the other three serotypes, is shown. Studies of hundreds of virus strains showed that for DENV-2, there were four distinct genotypic groups (simplified here) that correlated with the geographic origins of the patient samples, with sequence variations of about 6 percent or more (Rico-Hesse, 2007). Here we have highlighted two genotypic groups, the American and Southeast Asian (SE Asian) genotypes because these groups differed in their clinical disease presentations; that is, samples from patients with DF fell into all genotypes, but those from DHF patients fell into only one of the genotypes (SE Asian).
The American genotype had been circulating in the American continent before the 1980s, without causing major epidemics or severe disease (DHF); in 1979–1980, the SE Asian genotype was introduced, most probably via returning Cuban military personnel who had been infected in Vietnam, after the war (Rico-Hesse et al., 1997). This SE Asian genotype virus introduction was directly associated with the first occurrences of DHF in the Americas, in spite of having numerous serotypes circulating before this time; this genotype, with its high virulence characteristic has since spread to the rest of the world, and has seemed to displace the native genotypes in other continents also (e.g., Africa) (Messina et al., 2014).
In our case, the American genotype viruses have not been isolated on this continent since 1995, in northern Mexico (see bottom black highlight in Figure A10-1) and in northern Peru, in 2000 (Cruz et al., 2013), and the viruses isolated on the Mexican border with Texas, in 2005, are now SE Asian genotype viruses (see top black highlight in Figure A10-1). Thus, a virus variant that evolved within serotype 2 (maintaining its antigenic structure) became more virulent to humans and was more transmissible than the variants native to this and other continents. Unfortunately, because we had no animal models of disease at
SOURCE: Rico-Hesse, 2007. Reproduced with permission from Oxford University Press, on behalf of Clinical Infectious Diseases.
the time, we could not prove the direct link of infection by the SE Asian genotype to higher virulence and transmission.
This same effect, of infection by a specific genotype and more severe disease and transmission, has been shown for a Sri Lanka variant of DENV serotype 3 (Messer et al., 2002, 2003), but has not yet been demonstrated for genotypes of DENV serotypes 1 and 4. It is also important to point out that several reports have suggested that DENV was undergoing intragenomic recombination events, with the production of virions with nucleotide or antigenic properties of several genotypes or serotypes (Holmes et al., 1999; Holmes, 2006), but these studies were shown to be a result of technical errors in sequencing methods, and subsequent errors in the GenBank sequence postings. Therefore, the more virulent genotypes we discuss here are a result of independent mutation events that have led to the production of distinct virus lineages that can outcompete other members of the same antigenic groups (DENV-2 and DENV-3), and they have spread throughout the world. However, we still have not determined the exact parts of the virus genome that lead to these epidemiologic or virulence characteristics. Our studies with infectious clones and recombinant laboratory techniques have allowed us to pinpoint some parts of the virus that we believe can lead to increases in replication that translate into higher virion production, increased mosquito infectivity and vector competence, and increased human virulence and pathogenesis.
Virus replication in target cells Owing to the lack of animal models of disease, and only indirectly, by observation of human clinical presentations (during prospective epidemiologic studies), it has been very difficult to measure which factors lead to increased DENV replication and subsequent disease. If we focus on the production of DF, and not on the complex and little understood effects of our own immune system in causing DHF, we can measure differences in replication abilities of DENV in their primary target cells, taken directly from uninfected individuals. Some human tropism studies in biopsied materials from patients with or without DF, and some autopsy specimens from those dying of DHF, had shown that dendritic cells and macrophages were the first cells to be infected and replicated DENV at higher rates (Wu et al., 2000; Jessie et al., 2004). However, because of ethical reasons, there have been very few early studies of what happens to humans right after being infected by mosquito bite (Sabin, 1952). Some of these studies are being analyzed in more detail (Snow et al., 2014), but there are too few subjects and too many variables to be able to identify specific factors such as the exact dose of DENV needed to produce disease. Also, these “volunteers” were not biopsied or analyzed pathologically to determine the exact events leading to the sites of DENV replication after infection. In our laboratory, we measured the replication abilities of DENV of different genotypes, in monocyte-derived dendritic cells (DCs) from unidentified blood donors (Cologna et al., 2005).
In these cells, cultured in vitro (or ex vivo), we controlled for the numbers of cells and the dose of DENV used to infect, and we compared the infection and production abilities of 19 low-passaged (less than 4 passages from the patient’s sample) virus strains, representing the SE Asian (n = 12) and American (n = 7) genotypes of DENV-2 (see Figure A10-2). First we measured the number of cells infected by each genotype (by detection of expressed viral protein) (see Figure A10-2A), and then the number of virions produced by the same cells (using RNA genomes as a surrogate) (see Figure A10-2B). Surprisingly, the number of cells infected was higher for the less virulent, American genotype strains, and this varied by blood donor. However, when the numbers of viral RNA genomes (DENV are notoriously difficult to measure as infectious particles) were compared across donors, all donor samples produced much higher amounts of SE Asian genotype viruses. This means that American viruses may infect more target cells, but they produce less progeny viruses after a single round of replication (at 48 hours post-infection), and that some donors (not previously exposed to DENV) have some innate, probably genetic, cellular factors that prevent DENV replication. These differences in replication ability are independent of any measurable immune system response (adaptive immunity), since we are growing them in purified DC cultures. Therefore, segregation of DENV strains into nucleotide variant groups, or genotypes, helps us define evolutionary differences that increase the probability of the virus to infect human cells, produce viremia, and cause disease.
DENV determinants of replication To study the specific structures of the DENV RNA genome or expressed proteins that might determine differences in replication ability, we constructed chimeras of SE Asian and American genotype viruses, using recombinant DNA techniques (Cologna and Rico-Hesse, 2003). The sites for possible replication and/or virulence determinants were selected first by complete genome nucleotide sequencing of six SE Asian genotype strains and six American genotype strains (Leitmeyer et al., 1999). Their viral RNA nucleotide sequences were determined by chemical analysis (primer-extension Sanger sequencing off of viral RNA, with no cloning) from patient serum samples, so as not to introduce mutations during passage in cells, either by biological or enzymatic amplification techniques.
When these sequences were aligned, we found consistent differences across many different areas of the genome, but only one difference in an encoded amino acid, at E390 (N to D), which changed the charge and thus probably the structure of the proteins on the outside of the DENV virion. However, two other consistent differences occurred at sites in the untranslated parts of the genome, in the 5’ UTR (two nucleotides) and in the 3’ UTR (14 nucleotides, including 10 deletions) that we hypothesized could significantly alter the folding patterns of these RNA regions, thus, altering their ability to initiate or control replication and possibly switch to template translation, in order to control virion production.
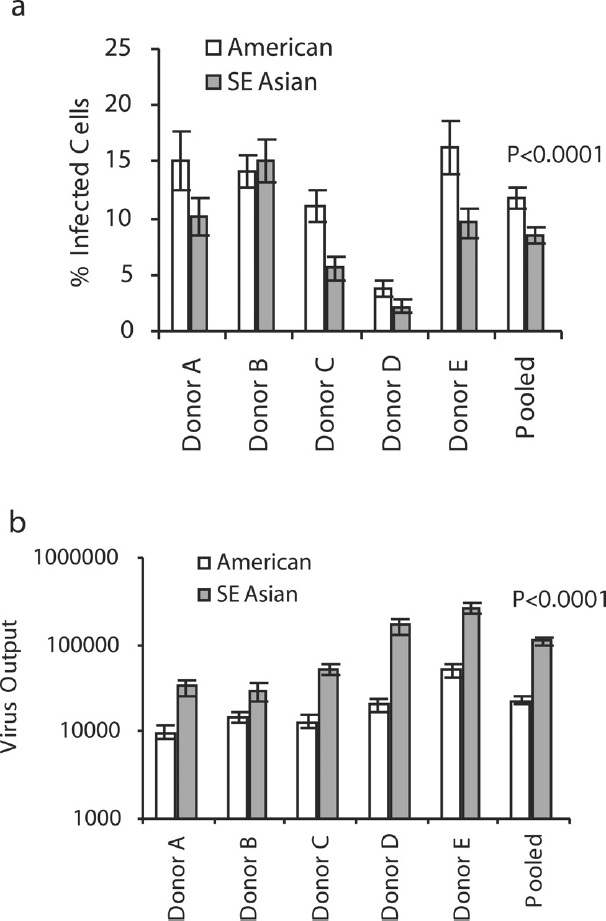
NOTE: Within each graph, the white bars represent the mean results of seven American genotype viruses, and the gray bars represent the results from 12 SE Asian genotype viruses. Error bars are SE, with infections in triplicate.
SOURCE: Cologna et al., 2005. Copyright © 2005, American Society for Microbiology. All Rights Reserved. Reproduced with permission from ASM Journals.
We later expanded these in silico studies, to determine the RNA folding patterns of viruses from all four genotypes, and found that strains from each genotype of DENV-2 had differing degrees of complexity in their folding patterns (see Figure A10-3), including overlaps (or pseudoknots), and these patterns seemed to correlate with rates of replication and/or virulence (Rico-Hesse, 2009). This suggested that these untranslated portions of the genome could in fact directly control initiation of replication, especially since they are the binding sites for
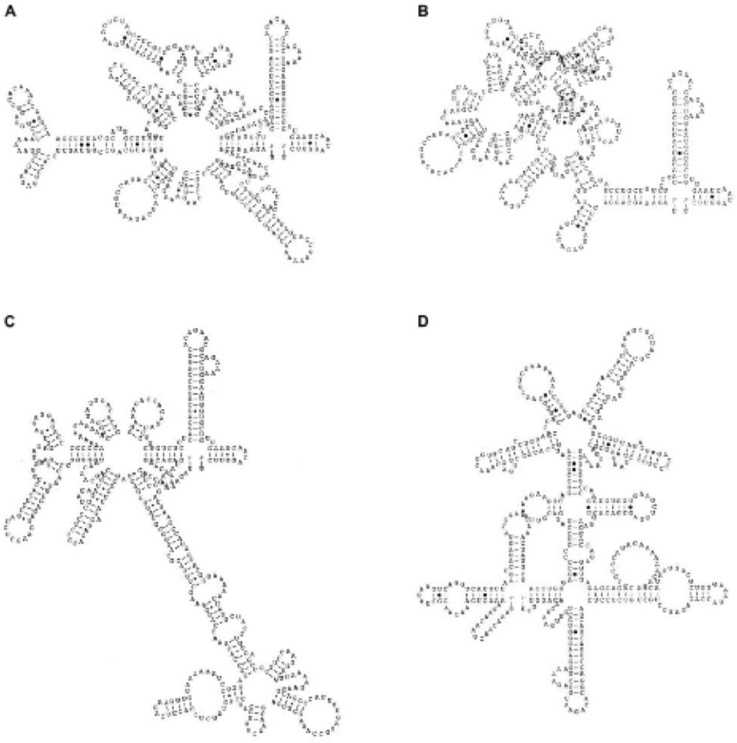
SOURCE: Rico-Hesse, 2009. Reproduced with permission from Future Medicine Ltd.
RNA polymerase, are responsible for genome circularization, to form replication intermediates (dsRNA), and for initiation of translation of the DENV polyprotein, thus switching from RNA replication to virion production. These hypotheses are currently being evaluated using novel and complex approaches in cell biology, viral chimeras, and biochemical and physical structure analyses.
Our studies with chimeras of SE Asian and American genotype viruses, with substitutions in the 5’UTR, EN390D amino acids, and 3’UTR, demonstrated that all three of these sites are important determinants of virus replication in primary human cell cultures (macrophages and DCs) (Cologna and Rico-Hesse, 2003). We inserted these American genotype sites into an infectious background clone of a SE Asian genotype virus, showing that replication and output rates were similar to wild-type American genotype virus in human cells. It remains for us to do the reverse experiment, by inserting SE Asian sites into an American genotype virus background to determine if replication and output can reach those of wild-type SE Asian viruses. However, modification of these structures did not affect their growth in C6/36 mosquito cell cultures. It should be noted that the C6/36 cell line is a single cell type and therefore does not represent the whole mosquito; these cell lines are known to be defective in some innate, cellular immune responses and therefore may not represent virus tropism in mosquitoes.
In 2003, when these studies were performed, the emphasis was placed on the exterior proteins of the virus as determinants of tropism and replication, meaning viral attachment sites on host cells could control the eventual output of virus. But, as we mentioned above, the number of infected cells does not actually determine the output of virus for the DENV-2 viruses we have studied. In fact, these studies helped us focus further on the role of the 3’UTR, its folding pseudoknots, and cellular factors for controlling levels of virion production from infected human cells. This is currently a very novel concept in the areas of virus replication and pathogenicity.
DENV determinants of virulence in humanized mice In 2005 we reported on the first animal model of DENV disease (DF), where NOD/SCID (immunodeficient) mice that were engrafted with hematopoietic stem cells derived from human umbilical cord blood (CB-hu-mice) were infected by injection of one DENV-2 strain, K0049 (low passage, SE Asian genotype). These mice developed viremia and the same clinical signs of disease as humans (fever, thrombocytopenia, erythema) (Bente et al., 2005). These humanized mice develop many components of the human immune system, including macrophages, dendritic cells, mast cells, and some lymphocytes (B and T cells), but they do not make specific antibodies to the infecting virus, nor do they produce educated T cells (required for DHF). Figure A10-4 outlines the procedures used to prepare these CB-hu-mice. Many other types of humanized mice have since been developed, and this is a rapidly changing field of research (for review see Brehm et al., 2013).
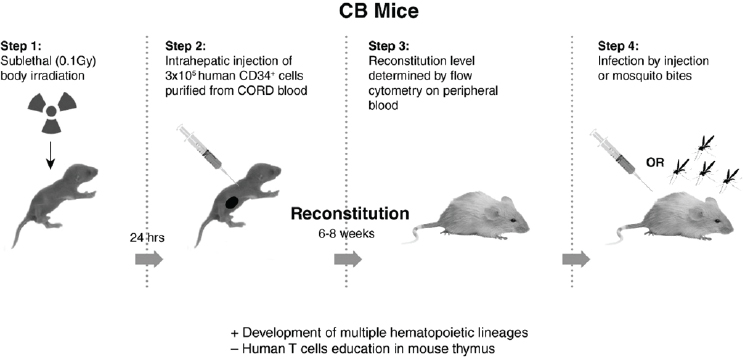
NOTE: Advantages (+) over other mouse models, factors missing (–) compared to other humanized mice.
After our initial report, we tested injection of eight different DENV-2 viruses (106 PFU equivalents, subcutaneously), representing the four genotypes, to demonstrate consistent differences in virulence of these viruses, in humanized mice (Mota and Rico-Hesse, 2009). However, we used a newer, more immunodeficient strain of mice, called NOD/SCID/IL2r gamma null (NSG), as recipients of the human umbilical cord blood stem cells (Figure A10-4), as these proved to be much better at attaining higher engraftment levels, and are currently still being used by many investigators attempting to study human-restricted pathogens. As can be seen in Figure A10-5, the representative viruses we selected produced statistically distinct viremias for each of the virus genotypes, with the SE Asian viruses highest and longest viremias, and the American genotype viruses lower (see Figure A10-5B), but not the shortest viremias (West African sylvatic genotype, Figure A10-5D), thus supporting our previous studies of epidemiologic relationships and growth in primary human target cells.
Other clinical signs were notably different, depending on the infecting DENV genotype, and statistical differences could be seen in fever, erythema, and thrombocytopenia levels. In addition, we performed studies of virus tropism, with a SE Asian genotype virus, demonstrating replication in numerous human cells, by flow cytometry and immunohistochemistry. These studies showed DENV replication in differentiated human B cells and in numerous, unidentified cells in the humanized mouse bone marrow (Mota and Rico-Hesse, 2011). It remains to be seen if we can identify all of the different types of human cells infected in these mice, as the numbers of these cells may be too small to enumerate, including those in lymph nodes, which are atrophied in these immunodeficient mice.
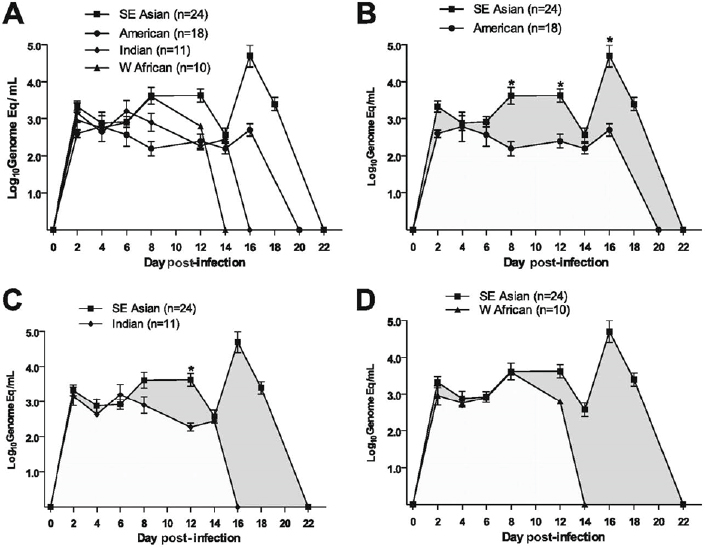
NOTE: Error bars are SE, and n = number of infected mice per virus genotype.
SOURCE: Mota and Rico-Hesse, 2009. Reproduced with permission from American Society for Microbiology.
Further improvements of this animal model may lead to a better understanding of DENV tropism for human cells, and their contribution to pathogenesis of DF and DHF.
DENV determinants of pathogenesis, including mosquito saliva To further develop the humanized mouse model of DENV disease, we initiated studies using only one strain of DENV-2, K0049, from the most virulent, SE Asian genotype, but this time by infecting the CB-hu-mice by the natural route of infection, mosquito bite (Cox et al., 2012). This entails inoculating (with very small amounts of virus, < 40 PFU eq.) female Aedes aegypti mosquitoes reared in the laboratory, keeping them in a biosafety level-3 lab facility for the extrinsic incubation period (7–9 days), and having them bite mouse footpads (best mimics of human epithelium), in order to transmit the virus via saliva.
These studies are extremely difficult to perform, as they require special containment facilities for both immunodeficient mice and mosquitoes, and then working with infectious, live mosquitoes in the BSL3. These studies also required titrations of how many mosquitoes are needed to bite each CB-hu-mouse, to get 100 percent development of DF; the final tally was four mosquitoes per mouse. However, our results were extremely novel, and not only did the mice have higher and more extended viremias when infected via mosquito bite (Figure A10-6), but they were able to make specific antibodies to the virus (IgM only; no class switching without T cells), and even to the mosquito saliva proteins. The erythema and thrombocytopenia were also higher, suggesting that the mosquito bite delivery has extreme effects on the virus as an antigen (opsonization), on hemostasis (probably on platelets and endothelial cells also), and these mice cannot clear the virus with their deficient immune system until after 54 days.
There are many factors that remain to be tested in CB-hu-mice, including the role of specific mosquito saliva proteins on the effects to virus and host cells, and the innate immune response, but fortunately, these mice are ready to be used in
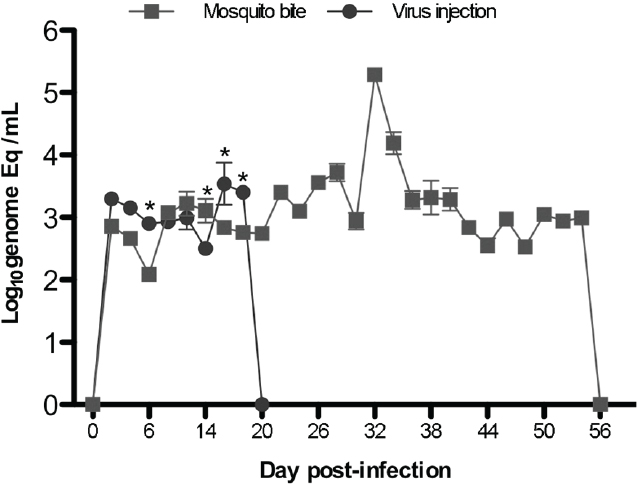
SOURCE: Cox et al., 2012. Reproduced with permission from American Society for Microbiology.
tests of antivirals or other treatments, to suppress DF clinical disease. However, these results also put into question the validity of any animal tests that use injection of virus, without the mosquito saliva components that occur during natural DENV transmission.
DENV determinants of mosquito infection and transmission We began studies of DENV-2 strain replication, dissemination, and transmission dynamics in Aedes aegypti mosquitoes in the late 1990s in parallel with virus virulence studies (Armstrong and Rico-Hesse, 2001). In the case of DENV, this species of mosquito has been proven to be more competent than the Aedes albopictus mosquito, although the latter was the main vector of DENV epidemics in Hawaii and in several countries along the Mediterranean. Aedes aegypti populations (collected in the field) have also been shown to differ genetically and show differences in vector competence, for differing DENV strains (Armstrong and Rico-Hesse, 2003).
Our studies have shown that although DENV-2 of the four different genotypes bind to mosquito midguts at the same rates (Cox et al., 2011), their levels of replication and dissemination are extremely varied, thus suggesting that viral infection via receptors on the mosquito midgut is not the constraining factor for virus dissemination, but rather, the rates of replication and virion production in mosquito cells. In fact, for SE Asian and American genotype viruses in low passage colonies (F < 4) of mosquitoes established from the field (McAllen, Texas), there is an up to 60-fold difference in the potential for these mosquitoes to be able to transmit virus (SE Asian >>American). See Figure A10-7 for a comparison of vectorial capacity (Anderson and Rico-Hesse, 2006).
When we infected these mosquitoes orally (by imbibing blood in a chamber), with both SE Asian and American viruses in the same amounts in spiked blood, as a direct competition experiment, the SE Asian viruses were able to disseminate into the salivary glands by day 7 versus day 10 for American genotype viruses, in mosquitoes from varied geographical collection sites (Cologna et al., 2005). This means that the SE Asian viruses have an exponentially higher degree of transmission efficiency over American genotype viruses, and this could explain their more recent displacement of the native genotype viruses on several continents. Thus, specific DENV-2 variants have evolved and adapted to cause more viremia in human hosts (virulence) and to be more infectious and transmitted at much higher rates by their main mosquito vector (vector competence), which could explain their changing patterns of infection and transmission around the world.
These results suggest that we should better prepare for DENV emergence by focusing control efforts on those viruses that have been shown to belong to the genotypes that show increased virulence and transmission characteristics (SE Asian for DENV-2; Sri Lankan for DENV-3). We should also focus our attention on these variants to understand the complex interaction between infection and pathogenesis in the human host and to better devise vaccines and treatments that would protect against the most dangerous genotypes.
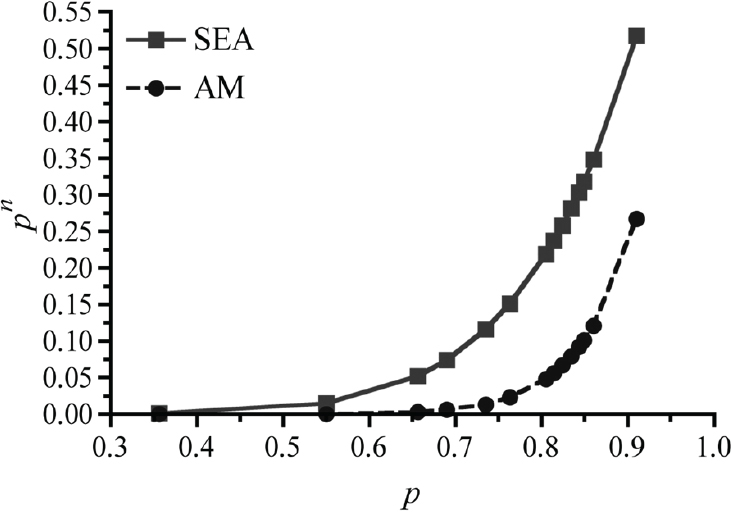
SOURCE: Anderson and Rico-Hesse, 2006. Reproduced with permission from American Journal of Tropical Medicine and Hygiene.
Chikungunya Viruses
Disease Characteristics and Viral Genetic Variability
The recent introduction and establishment of transmission of CHIKV in the Americas, first detected in December 2013, has made the study of this virus a priority for our nation. So far, autochthonous transmission has been demonstrated in Florida, and it is suspected that this virus will establish itself soon in the remainder of the U.S. Gulf Coast. Historically, French scientists had been the only ones prioritizing research on this virus, mainly because their overseas laboratories (Pasteur Institutes) were located in areas of endemic CHIKV transmission, and because many of their citizens were returning sick from vacationing in their former colonies (for review see Thiberville et al., 2013). However, the disease caused by this virus, chikungunya fever, and its related, severe arthralgias, have now become a concern to most of the developed countries, due to rapid travel
and introduction of viremic humans to even temperate climates, where the main vectors of CHIKV are present, both Aedes aegypti and Aedes albopictus.
The main problem with dealing with these two efficient viral vectors is that these mosquitoes differ in their biting habits (albopictus bites other animals in addition to humans), their habitat (albopictus can live farther away from urban areas), and their tolerance to low temperatures (infected albopictus eggs can overwinter in some of the coldest climates). In addition, the virus itself differs immensely from DENV, in that it infects other parts of the human body, including keratinocytes and fibroblasts, but causes almost identical symptoms and signs of disease as DENV, which make its extremely difficult to distinguish these two diseases. But more importantly, CHIKV causes severe, long lasting, and debilitating arthralgias in children and older adults, and it has an extremely high attack rate of 90 percent, which means it causes disease in 9 out of 10 infected individuals (versus 10 percent for DENV) (Schwartz and Albert, 2010).
Although there is only one serotype of virus, and infection produces lifelong protection against reinfection, the majority of the world’s population is currently susceptible to CHIKV, and this virus can be transmitted by mosquitoes that live in most parts of the globe. Therefore, we expect CHIKV to become the most prevalent and important emerging arbovirus of our lifetime.
Phylogenetic Trees and Structures
The CHIKV genome consists of approximately 12,000 nucleotides of RNA, which encode 9 proteins, in the order 5′-nonstructural proteins 1-4-capsid-envelope proteins1-3–protein 6k-3′; in addition, there are two UTRs on each end of around 75 nucleotides each, and a poly(A) tail on the 3′ end. Numerous studies have used comparison of nucleotide sequences from either the E2 or E1 envelope genes to generate phylogenetic trees of evolutionary relationships of CHIKV strains from around the world (Lanciotti et al., 1998; Powers et al., 2000). The most recent phylogenies, using complete coding genome sequences, consistently classify CHIKV into three genotypes that correspond with geographic origin: the West African, Asian, and Eastern Central Southern African (ECSA) (Thiberville et al., 2013). These investigators have also derived routes of spread of these genotypes, and most seem to have emerged from West Africa in the 1950s and 1960s, to other African regions (ECSA) and Asia, where independent cycles of evolution formed the new genotypes.
So far, most researchers have been concerned about the recent expansion of the ECSA genotype into islands in the Indian Ocean and from there to the Indian subcontinent, causing massive epidemics in 2004–2008. These events prompted concern around the world, with the first major funding initiatives to produce CHIKV vaccines and antivirals, and to investigate the viral biology and structural determinants of virulence. In contrast with DENV, where numerous structures of virions at various stages of maturation and resolution have been determined, and
from all four serotypes, for CHIKV there is no available complete virion structure (only for a virus-like-particle) (Sun et al., 2013).
We have shown this here, in Figure A10-8, with some of the purported sites of biological significance highlighted on the glycoprotein envelope protein (E1, E2, E3) trimer structure, as described by others (Voss et al., 2010). By using these predicted virion structures as a backdrop, we can see that three of the most important virus neutralization, virulence, and mosquito transmission determinants are all exposed on the end of the glycoprotein trimer (Figure A10-8B). Unfortunately, for DENV, the structures described to date do not show simple, consistent sites for some of these biological characteristics, and this may be complicated by the fact that those viruses show such varied antigenic structure (serotypes) and virulence characteristics.
Virus Replication in Target Cells
Although numerous reports describe growth of different strains of CHIKV in keratinocytes and fibroblasts in culture, a recent report disputes those results, and concludes that keratinocytes first serve as an antiviral defense on the skin, with specific innate immune factors secreted (Bernard et al., 2014). However, there are ample indications that fibroblasts, epithelial, and endothelial cells are infected and produce virus progeny soon after infection. In fact, these cells are probably producing the majority of the inflammatory response in joints and muscles, as they produce immune factors of activation and recruitment such as cytokines and chemokines (Sourisseau et al., 2007). In addition, because primary infections by any CHIKV strain seem to induce lifelong immunity to disease, it is most probable that patients make very high amounts of antibodies (and specific T cells and plasmablasts) that can serve to neutralize any incoming virus during a secondary infection. Such a strong neutralizing epitope has been described in a monoclonal antibody derived from patient’s plasma, and its primary binding site on the virion has been defined on E2, across the I121 and W64 amino acids (see Figure A10-8B).
The current problem with understanding the complete cycle of CHIKV replication and disease causation is that there are no detectable infectious virus particles in the joints or other organs of animals (mice and nonhuman primates) infected and studied after viremia disappearance, during the expected chronic sequellae. Immunodeficient and very young mice (Couderc et al., 2008) and some primates (Labadie et al., 2010) have been shown to develop CHIKV disease similar to humans, but none of these models fully mimics the route of inoculation and complete pathological picture as in humans.
The most recent study, in wild-type mice, suggests that a single amino acid on the E2 glycoprotein (E282), and some epistatic site differences (see Figure A10-8B), may determine the amount of dissemination and arteritis in this model; however, these mice do not entirely reflect the pathological processes
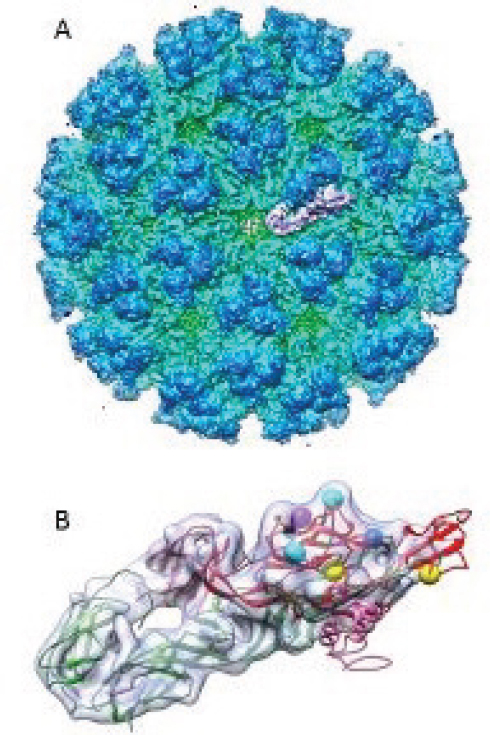
SOURCE: Cryo electron density map modified from Sun et al., 2013 (EMDB-5577); X-ray crystal structure was modified from Voss et al., 2010 (PDB-3N42).
occurring in humans, as they develop footpad and hind limb swelling only, without showing clinical signs (viremia, rash, fever, weight loss) and recumbence as in humans (Ashbrook et al., 2014). Thus, we are currently at a lack of complete animal models in which to study the pathogenesis of CHIKV and DENV, including the immune mechanisms and factors that might lead to severe disease presentations.
CHIKV Determinants of Pathogenesis, Including Mosquito Saliva
We propose to use a newer—although more complicated—type of humanized mouse to study immunopathogenesis of both DENV and CHIKV to attempt to develop disease that fully mimics human signs and postviremia induction of severe sequellae (DHF with secondary dengue, arteritis/arthritis in chikungunya fever). The preparation of these mice, known as bone marrow-liver-thymus humanized mice (BLT-hu-mice), is outlined in Figure A10-9. In this case, the adult NSG mice, the same strain as used for CB-hu-mice, are surgically implanted (suprarenal capsule) with human stem cells derived from fetal liver and thymus, for a much more complete reconstitution of the human immune system, including adaptive immunity (for a review see Brehm et al., 2013). These mice attain high levels of engraftment of the mouse bone marrow, substituting human cells, and because of the presence of human thymus, they are able to continually produce functional B and T cells, and thus make specific human antibodies, with class switching (IgM and IgG antibodies).
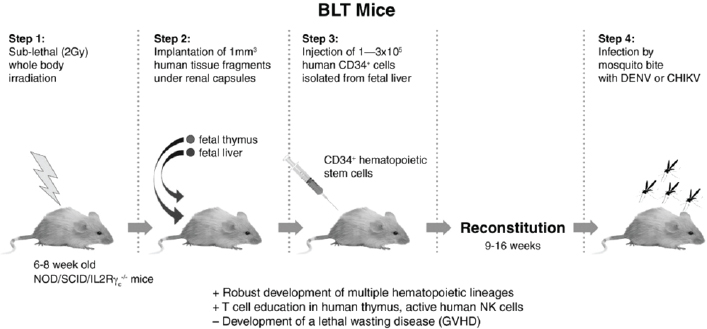
NOTE: Advantages (+) over other mouse models; disadvantages (-) compared to other humanized mice.
Although these mice are extremely difficult to produce, they theoretically allow for the measurement of human immune responses, including innate and adaptive factors, which could possibly lead to the models necessary for studying severe DENV and CHIKV disease. These animals could also potentially be used to determine the effects of mosquito saliva proteins, as these have been reported to be important mediators of infection by CHIKV, in cell culture and in wild-type mice (Thangamani et al., 2010). Use of these mice could allow us to perform natural route of infection experiments with CHIKV, which we have shown drastically modify the effects of infection and immunity to DENV.
CHIKV Determinants of Mosquito Infection and Transmission
Previous studies using infectious clones of CHIKV (ECSA genotype) and their recombinant modifications implicated a site in the E1 glycoprotein (A226V) as a determinant of switching vector competence from Aedes aegypti to Aedes albopictus (Tsetsarkin et al., 2007, 2009). This would have created a very significant problem for the expansion of CHIKV disease into entirely new areas and new human populations, or a new mode of vector adaptation. However, more recent experiments using over 35 different colonies of Aedes albopictus and Aedes aegypti mosquito strains, and testing CHIKV strains with the E1226A and E1226V mutation in the ECSA genotype backgrounds, and the new, Asian genotype virus that has been introduced recently to the Americas, have not shown consistent statistically significant differences in vector competence for either species of mosquito (three groups showed lower dissemination in albopictus) (Vega-Rua et al., 2014). However, these studies failed to include sufficient Asian genotype virus strains to fully evaluate their rates of transmission by either mosquito species, and these are the CHIKV genotype variants that concern us most now.
Another concern is that there is evidence that Aedes albopictus can replicate and transmit both DENV and CHIKV simultaneously, after oral infection (Vazeille et al., 2010); therefore, we need to evaluate the dynamics of transmission and virulence when both viruses are infecting mosquitoes and human hosts simultaneously. This adds a major degree of complexity to the measurements of evolution and virulence that should be taken into account for various assay systems and ultimately, in the way that arbovirus dynamics and emergence mechanisms can be modeled effectively.
Acknowledgments
Funding was provided by NIH, P41GM103832 (Chiu/CWH), Robert Welch Foundation, Q1242 (Chiu/CWH), NIH R01AI099483 (RRH), and NIH R01AI098715 (RRH).
References
Anderson, J. R., and R. Rico-Hesse. 2006. Aedes aegypti vectorial capacity is determined by the infecting genotype of dengue virus. American Journal of Tropical Medicine Hygiene 75(5):886-892.
Armstrong, P. M., and R. Rico-Hesse. 2001. Differential susceptibility of Aedes aegypti to infection by the American and Southeast Asian genotypes of dengue type 2 virus. Vector Borne Zoonotic Diseases 1(2):159-168.
Armstrong, P. M., and R. Rico-Hesse. 2003. Efficiency of dengue serotype 2 virus strains to infect and disseminate in Aedes aegypti. American Journal of Tropical Medicine and Hygiene 68(5):539-544.
Ashbrook, A. W., K. S. Burrack, L. A. Silva, S. A. Montgomery, M. T. Heise, T. E. Morrison, and T. S. Dermody. 2014. Residue 82 of the chikungunya virus E2 attachment protein modulates viral dissemination and arthritis in mice. Journal of Virology 88(21):12180-12192.
Bente, D. A., M. W. Melkus, J. V. Garcia, and R. Rico-Hesse. 2005. Dengue fever in humanized NOD/SCID mice. Journal of Virology 79(21):13797-13799.
Bernard, E., R. Hamel, A. Neyret, P. Ekchariyawat, J. P. Moles, G. Simmons, N. Chazal, P. Despres, D. Misse, and L. Briant. 2014. Human keratinocytes restrict chikungunya virus replication at a post-fusion step. Virology 476C:1-10.
Bhatt, S., P. W. Gething, O. J. Brady, J. P. Messina, A. W. Farlow, C. L. Moyes, J. M. Drake, et al. 2013. The global distribution and burden of dengue. Nature 496(7446):504-507.
Brehm, M. A., N. Jouvet, D. L. Greiner, and L. D. Shultz. 2013. Humanized mice for the study of infectious diseases. Current Opinions in Immunology 25(4):428-435.
Cologna, R., and R. Rico-Hesse. 2003. American genotype structures decrease dengue virus output from human monocytes and dendritic cells. Journal of Virology 77(7):3929-3938.
Cologna, R., P. M. Armstrong, and R. Rico-Hesse. 2005. Selection for virulent dengue viruses occurs in humans and mosquitoes. Journal of Virology 79(2):853-859. doi: 10.1128/JVI.79.2.853-859.2005.
Couderc, T., F. Chretien, C. Schilte, O. Disson, M. Brigitte, F. Guivel-Benhassine, Y. Touret, G. Barau, N. Cayet, I Schuffenecker, P. Despres, F. Arenzana-Seisdedos, A. Michault, M.L. Albert, and M. Lecuit. 2008. A mouse model for chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathogens 4(2):e29.
Cox, J., H. E. Brown, and R. Rico-Hesse. 2011. Variation in vector competence for dengue viruses does not depend on mosquito midgut binding affinity. PLoS Neglected Tropical Diseases 5(5):e1172.
Cox, J., J. Mota, S. Sukupolvi-Petty, M. S. Diamond, and R. Rico-Hesse. 2012. Mosquito bite delivery of dengue virus enhances immunogenicity and pathogenesis in humanized mice. Journal of Virology 86(14):7637-7649.
Cruz, C. D., B. M. Forshey, D. S. Juarez, C. Guevara, M. Leguia, T. J. Kochel, and E. S. Halsey. 2013. Molecular epidemiology of American/Asian genotype DENV-2 in Peru. Infections Genetics, and Evolution 18:220-228.
Guzman, M. G., S. B. Halstead, H. Artsob, P. Buchy, J. Farrar, D. J. Gubler, E. Hunsperger, A. Kroeger, H. S. Margolis, E. Martinez, M. B. Nathan, J. L. Pelegrino, C. Simmons, S. Yoksan, and R. W. Peeling. 2010. Dengue: A continuing global threat. Nature Reviews Microbiology 8(12 Suppl):S7-S16.
Holmes, E. C. 2006. The evolutionary biology of dengue virus. Novartis Foundation Symposia 277:177-187; discussion 87-92, 251-253.
Holmes, E. C., M. Worobey, and A. Rambaut. 1999. Phylogenetic evidence for recombination in dengue virus. Molecular Biology and Evolution 16(3):405-409.
Jessie, K., M. Y. Fong, S. Devi, S. K. Lam, and K. T. Wong. 2004. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. Journal of Infectious Diseases 189(8):1411-1418.
Labadie, K., T. Larcher, C. Joubert, A. Mannioui, B. Delache, P. Brochard, L. Guigand, L. Guigand, L. Dubreil, P. Lebon, B. Verrier, X. de Lamballerie, A. Suhrbier, Y. Cherel, R. Le Grand, and P. Roques. 2010. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. Journal of Clinical Investigations 120(3):894-906.
Lanciotti, R. S., M. L. Ludwig, E. B. Rwaguma, J. J. Lutwama, T. M. Kram, N. Karabatsos, B. C. Cropp, and B. R. Miller. 1998. Emergence of epidemic O’nyong-nyong fever in Uganda after a 35-year absence: Genetic characterization of the virus. Virology 252(1):258-268.
Leitmeyer, K. C., D. W. Vaughn, D. M. Watts, R. Salas, I. Villalobos, Chacon de, C. Ramos, and R. Rico-Hesse. 1999. Dengue virus structural differences that correlate with pathogenesis. Journal of Virology 73(6):4738-4747.
Messer, W. B., U. T. Vitarana, K. Sivananthan, J. Elvtigala, L. D. Preethimala, R. Ramesh, N. Withana, D. J. Gubler, and A. M. De Silva. 2002. Epidemiology of dengue in Sri Lanka before and after the emergence of epidemic dengue hemorrhagic fever. American Journal of Tropical Medicine and Hygiene 66(6):765-773.
Messer, W. B., D. J. Gubler, E. Harris, K. Sivananthan, and A. M. de Silva. 2003. Emergence and global spread of a dengue serotype 3, subtype III virus. Emerging Infectious Diseases 9(7):800-809.
Messina, J. P., O. J. Brady, T. W. Scott, C. Zou, D. M. Pigott, K. A. Duda, S. Bhatt, L. Katzelnick, R. Howes, K. Battle, C. Simmons, and S. Hays. 2014. Global spread of dengue virus types: Mapping the 70 year history. Trends in Microbiology 22(3):138-146.
Mota, J., and R. Rico-Hesse. 2009. Humanized mice show clinical signs of dengue fever according to infecting virus genotype. Journal of Virology 83(17):8638-8645.
Mota, J., and R. Rico-Hesse. 2011. Dengue virus tropism in humanized mice recapitulates human dengue fever. PLoS One 6(6):e20762.
Powers, A. M., A. C. Brault, R. B. Tesh, and S. C. Weaver. 2000. Re-emergence of Chikungunya and O’nyong-nyong viruses: Evidence for distinct geographical lineages and distant evolutionary relationships. Journal of General Virology 81(Pt 2):471-479.
Rico-Hesse, R. 1990. Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virology 174(2):479-493.
Rico-Hesse, R. 2003. Microevolution and virulence of dengue viruses. Advances in Virus Research 59:315-341.
Rico-Hesse, R. 2007. Dengue virus evolution and virulence models. Clinical Infectious Diseases 44(11):1462-1466.
Rico-Hesse, R. 2009. Dengue virus markers of virulence and pathogenicity. Future Virology 4(6):581.
Rico-Hesse, R., L. M. Harrison, R. A. Salas, D. Tovar, A. Nisalak, C. Ramos, J. Boshell, M. T. de Mesa, R. M. Nogueira, and A. T. da Rosa. 1997. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology 230(2):244-251.
Rothman, A. L. 2011. Immunity to dengue virus: A tale of original antigenic sin and tropical cytokine storms. Nature Reviews Immunology 11(8):532-543.
Sabin, A. B. 1952. Research on dengue during World War II. American Journal of Tropical Medicine and Hygiene 1(1):30-50.
Schwartz, O., and M. L. Albert. 2010. Biology and pathogenesis of chikungunya virus. Nature Reviews of Microbiology 8(7):491-500.
Snow, G. E., B. Haaland, E. E. Ooi, and D. J. Gubler. 2014. Research on dengue during World War II revisited. American Journal of Tropical Medicine and Hygiene 91(6):1203-1217.
Sourisseau, M., C. Schilte, N. Casartelli, C. Trouillet, F. Guivel-Benhassine, D. Rudnicka, N. SolFoulon, et al. 2007. Characterization of reemerging chikungunya virus. PLoS Pathogens 3(6):e89.
Sun, Y., J. Yan, H. Mao, L. Zhang, Q. Lyu, Z. Wu, W. Zheng, C. Feng, and Y. Zhang. 2013. Characterization of the complete genome of chikungunya in Zhejiang, China, using a modified virus discovery method based on cDNA-AFLP. PLoS One 8(12):e83014.






















