
7
Organic Nutrient Chemistry and the Marine Microbiome
Daniel J. Repetaa,* and Rene M. Boiteaua,b
INTRODUCTION
Vast expanses of the ocean are characterized by extraordinarily low concentrations of nutrients, but, nevertheless, support vibrant communities of marine microbes. In aggregate, these communities drive many of the important elemental cycles that sustain life on Earth. At the cellular level, open ocean microbes have optimized their size and geometry, biochemical composition, and lifestyle to grow efficiently in an oligotrophic environment. Microbial communities are likewise organized to maximize nutrient and energy transfer between cells, and efficiently recycle organic carbon, nitrogen, phosphorus, and trace metals. Energy and nutrient transfer occurs across a broad range of spatial scales. Large-sized marine algae and bacteria support epibiont communities that are physically in contact, exchanging nutrients and energy across cell membranes; other communities, which are physically far apart, rely on the horizontal mixing of ocean currents or the vertical pull of gravity to transfer nutrient- and energy-containing organic matter.
Marine productivity is limited by the concentration and availability of essential nutrients—particularly nitrogen, phosphorus, and iron—which in the open ocean occur at nM to pM concentrations. Heterotrophic bacterial communities are likewise limited by available carbon substrates. Each year, more than 10 GT of dissolved organic carbon (DOC) is added to seawater by the activity of marine microbes. Most of this organic matter is respired immediately, cycling over timescales of hours to days. However, some escapes immediate remineralization and accumulates in the water column, where it slowly degrades over several years. DOC concentrations are 40-80 µM throughout the water column, while dissolved organic nitrogen (DON) and phosphorus (DOP) concentrations in the upper ocean euphotic zone are µM and 10-1,000 pM, respectively (Hansell et al., 2009; Torres-Valdes et al., 2009); dissolved iron concentrations in nutrient-limited regions reach 50-100 pM. One of the unsolved mysteries of marine microbial biogeochemistry is that while microbial communities are usually carbon, nitrogen, phosphorous, or iron limited, these microbes nevertheless inhabit an environment where DOC, DON, DOP, and organically bound iron are abundant.
It is widely believed that the accumulation of organic carbon, nitrogen, phosphorous, and iron in the water column is due to the chemical form of the organic compounds that make up marine dissolved organic matter
___________________
a Department of Marine Chemistry and Geochemistry, Woods Hole Oceanographic Institution.
b Earth and Biological Sciences Directorate, Pacific Northwest National Laboratory.
* Corresponding Author: drepeta@whoi.edu.
(DOM). This in turn has led to a vigorous effort to characterize DOM at the molecular level to understand the relationship between molecular structure and bioavailability; however, marine organic geochemists have had to surmount a number of formidable technical and operational challenges. DOM is an extremely complex mixture of organic molecules, consisting of at least tens to hundreds of thousands of unique compounds. Individual components of DOM therefore occur at pM and fM concentrations, often at the limit of what can be measured analytically. Even when the problems of detection and measurement can be overcome, many of the compounds present in DOM are not active in marine biogeochemical cycles. Rather, they are the refractory end-products of organic matter degradation. These refractory compounds persist in the ocean for several thousand years, and are chemically unlike the proteins, nucleic acids, lipids, and carbohydrates that constitute most cells. The presence of a high concentration of refractory background DOM therefore complicates all efforts to study the chemistry of the marine microbiome. Finally, most chemical analyses rely on some type of spectrometric characterization, which often requires isolating the analyte of interest. DOM concentrations in the ocean are typically 0.5-1.0 mg/L, about 10–4 times less than the concentration of salt. Although several techniques have been used to isolate DOM, even the most efficient isolation schemes are only able to recover up to 60-70% of total DOM. The composition of the remaining 30-40% is, therefore, largely unknown (Hansell and Carlson, 2015).
Even in the face of these challenges, marine organic geochemists are making rapid progress in understanding the chemistry of the marine microbiome. These advances have benefited from parallel developments in analytical chemistry, microbial isolation and culture techniques, and advances in microbial genomics, transcriptomics, and proteomics. The combination of all three approaches has proven to be quite powerful. Here, we highlight two aspects of organic phosphorus chemistry and trace metal cycling in the marine microbiome. In each study, advances in chemical analyses, microbial culture, and microbial genomics played key roles in understanding how microbial communities interact to facilitate nutrient cycling in the open ocean.
ORGANIC PHOSPHOROUS CYCLING WITHIN THE MARINE MICROBIOME
Phosphorus is used in the synthesis of nucleic acids, phospholipids, energy storage products like ATP, and a suite of other essential metabolites. Phosphorus is delivered to the ocean by atmospheric dust, continental river runoff, and groundwaters, and is removed by the deposition of marine sediments. Unlike nitrogen, which can be fixed by ocean microbes from N2, all phosphorus present in the ocean is derived from external sources. Most phosphorus occurs as inorganic phosphate, which has µM concentrations in the deep sea, but is only a few nM in surface waters due to rapid biological uptake and subsequent export on sinking particles. Upper ocean phosphorus speciation is therefore dominated by organic forms, which accumulate as byproducts of the intense cycling of organic matter within the euphotic zone. DOP concentrations in the upper water column are therefore more than 10 times the concentration of inorganic phosphate.
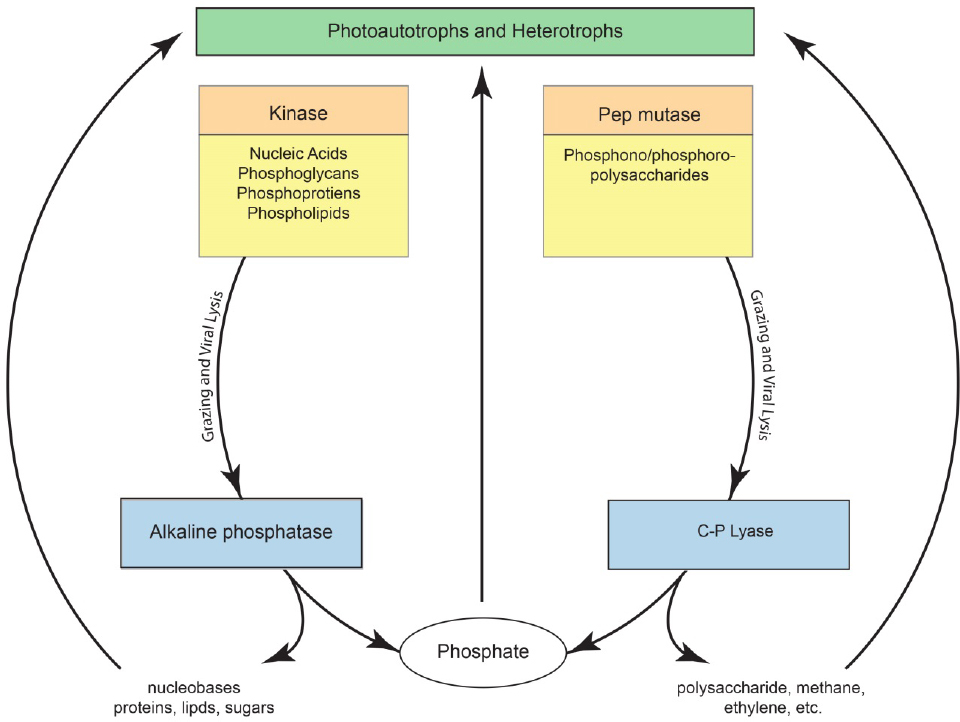
Inorganic phosphate is the preferred substrate of marine microbes. However, when phosphate concentrations are low, marine bacteria synthesize the enzyme alkaline phosphatase (APase), a relatively nonspecific phosphate monoester hydrolase that enables microbes to access many phosphorus-containing organic substrates (Karl, 2014). Metagenomic analyses suggest that approximately 50% of open ocean microbes can synthesize alkaline phosphatase (Sebastian and Ammerman, 2009). APase provides access to the large reservoir of organic phosphorus present in the upper water column, and numerous studies have shown a general inverse relationship between phosphate concentrations and APase activity (Torres-Valdes et al., 2009). When phosphate concentrations are high, APase activities are generally low, although there are important exceptions to this simple relationship. Nevertheless, most organic phosphorus produced by photoautotrophs is degraded by heterotrophic bacteria that release phosphate for further use. The production and degradation of organic phosphorus therefore links different microbial communities as shown in Figure 7-1.
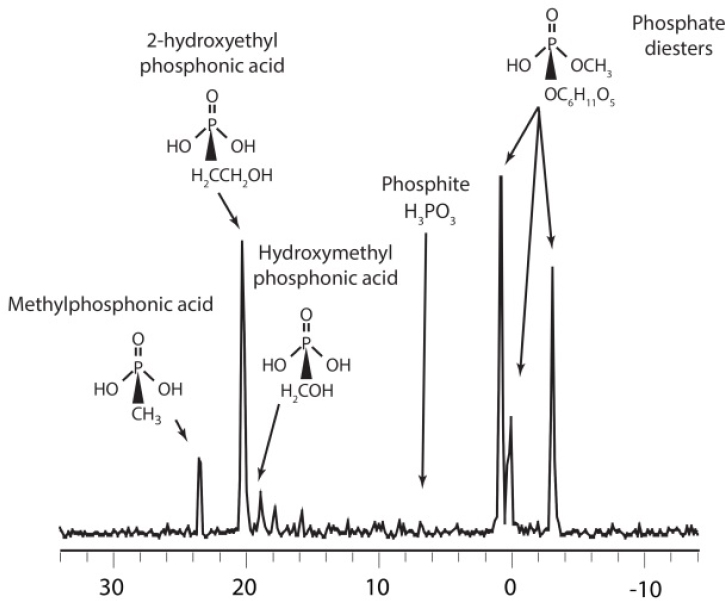
Common phosphorus-containing biochemicals like vitamins, nucleotides, and phospholipids have very low concentrations in seawater, typically only a few nM. The chemical identity of most DOP is therefore not known (Hansell, 2015). However, between 30% and 50% of DOP can be isolated by ultrafiltration, a technique that efficiently recovers high molecular weight (HMW)DOM with a molecular weight nominally >1 kD. Phosphorous-31 NMR spectra of HMWDOP show it to be a mixture of phosphate (70%), phosphonate (20%), and pyrophosphate
(10%) esters bound to a novel family of polysaccharides that are present throughout the ocean (Repeta et al., 2016). Further chemical analyses of HMWDOP show that phosphate esters are polysaccharide-methyl diesters, while the phosphonates are a mixture of esters including methylphosphonate, 2-hydroxyethylphosphonate, phosphite, as well as a suite of other phosphonates present in low abundance (see Figure 7-2).
Phosphonates have a carbon–phosphorus (C–P) bond, and the presence of phosphonates as 20% of HMWDOP is particularly striking. Phosphonate biosynthesis is restricted within microbial communities, and phosphonates do not appear to be major components of upper ocean microbial biomass. The abundance of phosphonates in DOP therefore results from strong selective preservation of phosphonate-containing polysaccharides due to as yet unrecognized molecular features. The C:N:P ratio of HMWDOM polysaccharide is ~300:20:1; a large fraction of organic nitrogen and phosphorus sequestered in the ocean is therefore stored within the same biochemical.
Phosphonates are synthesized by the rearrangement of phosphoenol pyruvate, a reaction catalyzed by the enzyme phosphoenolpyruvate mutase (PepM). PepM is present in ~7% of open ocean microbes, particularly within Proetobacteria, Firmicutes, Cyanobacteria, and Spirochaetes (Yu et al., 2013). Members of these phyla are among
the most widely distributed and abundant microbes in the ocean, and it is therefore not surprising that phosphonates are so ubiquitous in marine DOM. Phosphonates are not degraded by alkaline phosphatase, and preliminary experiments suggest that phosphate diesters and phosphonate within HMWDOM polysaccharides are likewise not hydrolyzed following treatment by APase. The resistance of DOP polysaccharide-methyl diesters to hydrolysis by APase may be an important reason why these organic compounds accumulate in the upper ocean. Phosphonates are degraded by the enzyme C–P lyase, which catalyzes the reduction of the organic compound and oxidation of phosphonate to phosphate. The products of C–P lyase activity on methylphosphonate and 2-hydroxyethylphosphonate are therefore methane, ethylene, and phosphate.
Nearly 50 years ago, geochemists first reported that the upper ocean is supersaturated with respect to the atmosphere in methane, a greenhouse gas that is produced by Archea under strictly anaerobic conditions. The presence of high concentrations of methane in well-oxygenated surface waters therefore posed a challenge to microbial biogeochemistry, which was posed as a “marine methane paradox.” Microbial degradation of methylphosphonate provides one solution to the methane paradox, in which methane is released as a byproduct of microbial cycling of HMWDOM phosphonates. Daily cycling of only 0.25% of HMWDOM inventory yields enough methane to support measured ocean-to-atmosphere fluxes of this greenhouse gas (see Figure 7-1).
Recent application of dilution-to-extinction cultivation techniques has allowed for the isolation and laboratory culture of marine bacteria with C–P lyase from seawater that can use model phosphonates like methylphosphonate and 2-hydroxyethylphosphonate, or HMWDOM as their sole source of phosphorus. Mutant strains of these same bacteria with a portion of the C–P lyase operon knocked out lose their ability to grow on HMWDOM, showing this
metabolic pathway is essential for HMWDOM phosphonate utilization. A metagenomic analysis of the distribution and abundance of C–P lyase within marine microbes has not been made, and it is unclear how much overlap there is between the microbial communities that express PepM and the communities that express C–P lyase. But, it is clear from existing data that these two communities are not identical: Some microbes can only synthesize phosphonates, while others can only degrade them. Complete phosphonate cycling may therefore require the activity of both communities, and the accumulation of HMWDOM may therefore arise from the imperfect coupling of phosphonate-producing microbes with PepM, and phosphonate-degrading microbes with C–P lyase.
SIDEROPHORES AND THE CYCLING OF ORGANICALLY BOUND IRON WITHIN THE MARINE MICROBIOME
Iron is an essential micronutrient for nearly every organism in the ocean. It is at the heart of cellular machinery that carries out photosynthesis, respiration, nitrate assimilation, and nitrogen fixation (Morel and Price, 2003). Each cell must assimilate enough iron to meet its needs, which can typically range from 0.2 to 20.0 mmol iron per mol phosphorus (Twining and Baines, 2013). In regions of the ocean where bioavailable iron is scarce relative to other nutrients, microbial growth can be iron limited. Surface waters across large regions of the Southern Ocean, eastern tropical Pacific Ocean, and western subarctic Pacific Ocean have large inventories of nitrate and phosphate unused by microbes due to an insufficient supply of iron (De Baar et al., 2005).
In the early 1990s, oceanographers demonstrated that changes in iron supply to these regions can affect the rate of microbial growth and carbon/nutrient export from surface waters. Such experiments led to a major paradigm shift about the importance of micronutrients in marine ecosystems. Since then, there has been a growing drive to understand the links between iron cycling and marine biogeochemistry. Hypotheses have been formulated to suggest that an increase in the supply of wind-borne iron to the Southern Ocean drove increased plankton growth and removal of carbon dioxide from the atmosphere during the Earth’s glaciations over the past several million years. Such work has also sparked interest, and controversy, over the feasibility of iron fertilization as a means to increase ecosystem productivity and sequester anthropogenic CO2. Fundamental insights into these issues rely on accurate knowledge of the rates of iron transformation and biological uptake in the ocean.
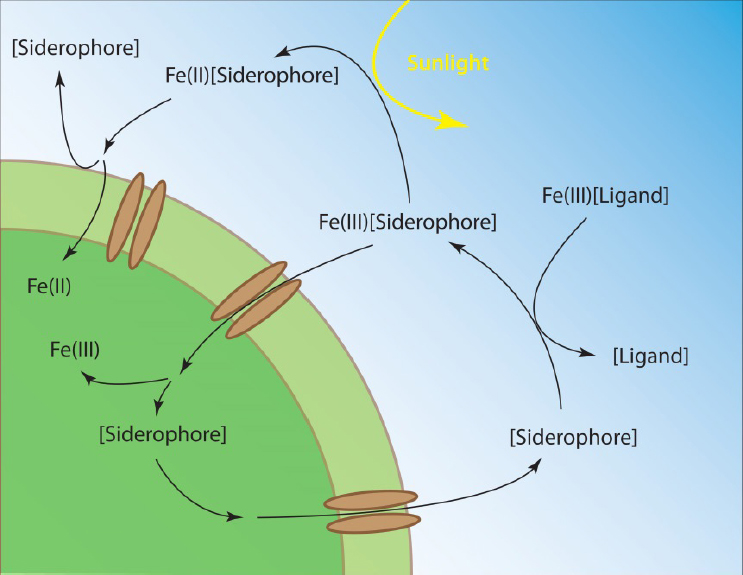
Nearly all bioavailable iron in the ocean is complexed by organic ligands of biological origin. Organic ligands prevent otherwise insoluble Fe(III) from precipitating as oxyhydroxides, increase the dissolution rate of iron from inorganic minerals and dust, stabilize iron released from hydrothermal vents and benthic sediments, and affect the rates of iron oxidative and reductive photochemical reactions (Gledhill and Buck, 2012; see Figure 7-3). Biological iron assimilation relies on the ability of microbes to access organically bound iron, for which several uptake strategies have evolved. In the simplest case, microbes can take up the free iron when it dissociates from organic ligand complexes. Some microbes, such as diatoms, can facilitate this process by reducing organically bound Fe(III) to Fe(II), which is readily taken up. Other microbes have specialized receptors capable of recognizing specific iron–ligand complexes, such as siderophores, and transporting them into the cell. Siderophores are strong iron-binding organic ligands that are exuded by microbes under conditions of scarcity to facilitate its uptake. These compounds compete with ambient iron ligands in the surrounding environment and complex it in a form that can be recognized and taken up by specific membrane receptors (Sandy and Butler, 2009; see Figure 7-3). The chemical composition and source of iron ligands therefore has important implications for iron reactivity and uptakes rates throughout the ocean.
Marine organic geochemists have sought to characterize iron-binding organic compounds in seawater and understand how these compounds mediate microbial uptake. Since dissolved organic carbon concentrations typically exceed dissolved iron concentrations by 4-6 orders of magnitude, characterization of organically chelated iron is therefore a formidable task that relies on identifying trace components within a complex and abundant organic matrix. A key advance in characterizing trace metal organic ligands in seawater was the coupling of advanced separation technologies, centered around ultra-high-performance liquid chromatography (HPLC) and high-resolution mass spectrometry, with data processing algorithms designed to search large amounts of data in ways that could quickly identify unique mass and abundance patterns imparted by trace metal isotopes. Current approaches use two complementary mass spectrometry methods: inductively coupled plasma mass spectrometry (ICPMS) and
high-resolution electrospray ionization mass spectrometry (ESIMS). ICPMS directly detects the metal of interest and provides a means to screen samples for the presence of organic–metal complexes. This approach has the major advantage that sensitivity is not dependent on the chemical species of the organic ligand, and therefore provides a quantitative measurement of each analyte (Boiteau and Repeta, 2015). However, ICPMS does not provide any information on the organic species. Once the retention time of the iron–organic complex has been determined by HPLC-ICPMS, ESIMS can be used to determine the mass and fragmentation pattern of the metal-containing compound and its chemical structure.
For iron, the approach leverages the exact mass difference and relative abundance of 56Fe (91.8%) and 54Fe (5.8%). When iron is incorporated into an organic compound, two isotopologs that reflect these mass and intensity differences are detected. Spectral processing algorithms quickly search the millions of ions collected during a typical sample analysis for those with the mass difference and abundance ratio of 56Fe and 54Fe, thereby identifying the iron ligands within the complex mixture of organic matter. This combination of analytical and computational methods allows iron and other trace metal–binding ligands to now be easily detected directly from seawater extracts (Mawji et al., 2008; Boiteau et al., 2016).
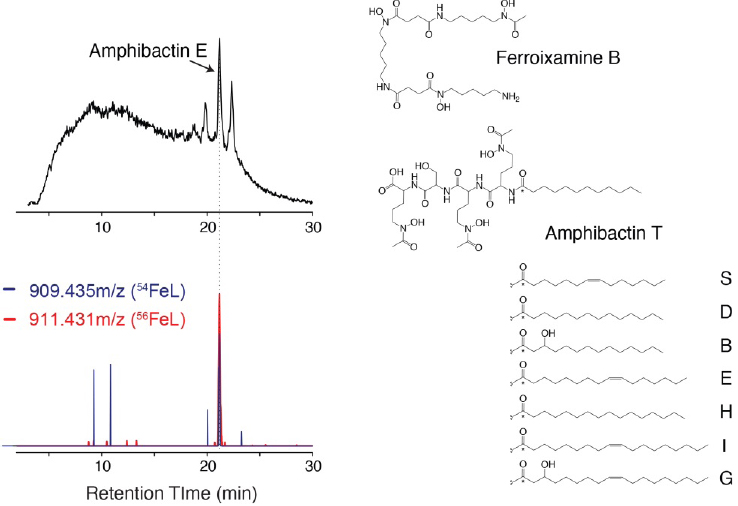
The application of advanced methods for the detection and identification of trace metal–organic complexes in natural seawater is just beginning, but existing data already show that the composition of trace metal ligands is highly dynamic across marine ecosystems. In the eastern tropical South Pacific Ocean, the composition of microbially produced siderophores changes across major nutrient regimes (Boiteau et al., 2016). Hydrophilic ferrioxamine siderophores were identified at low concentrations (1-2 pM) in nutrient-replete coastal waters and
macronutrient-depleted oligotrophic waters. In contrast, siderophore concentrations were up to fivefold higher in regions where iron concentrations were low, but macronutrient concentrations remained high. Increased concentrations of siderophores were accompanied by a dramatic shift in their composition from ferrioxamines to amphibactins—amphiphilic siderophores with high membrane affinity (see Figure 7-4). While all siderophores measured in high-iron inshore waters were bound to iron, nearly half the siderophores detected in low-iron waters were not metal bound. These uncomplexed siderophores were therefore available to capture iron that is remineralized during organic matter degradation, or which enters the ecosystem as atmospheric dust. As the amount and bioavailability of iron changes across the South Pacific Ocean, microbial strategies for acquiring iron change as well, impacting the concentration and distribution of siderophores.
Different enzymatic pathways direct siderophore production and the synthesis of their transporters that facilitate iron acquisition. Initial studies suggested that nonspecific and specific iron transporters were widely distributed in marine microbes (Toulza et al., 2012), but without a knowledge of siderophore composition, it was difficult to target and identify genes for siderophore synthesis. However, the direct measurement of siderophores in seawater has made the detection of siderophore synthesis genes in metagenomic catalogs much more feasible, and data are now beginning to appear that directly link iron speciation with the microbes that produce and take up siderophores. The high concentrations of amphibactins measured in the low-iron region of the eastern tropical South Pacific Ocean made amphibactin synthesis genes attractive targets for metagenome-based surveys of siderophore production. Amphibactins are synthesized by a pair of nonribosomal protein synthetase (NRPS) enzymes, which direct the assembly of the peptidic iron binding head group of amphibactins (Kem and Butler, 2015). Using amphibactin NRPS genes from several taxa of cultured Gammaproteobacteria as query sequences, we investigated the phylogeny
and spatial distribution of homologs within the Tara Oceans Metagenomic Catalogue. Numerous sequences with similarity to Vibrio NRPS enzymes were present in low-iron waters of the South Pacific and Atlantic Oceans, as well as in low-iron coastal regions off the western continental United States. Other measurements of radio-labeled iron uptake suggest that amphibactin-bound iron may be available to a wide variety of marine bacteria and, like phosphorus, microbial communities that synthesize and take up these organic nutrients might overlap, but may not be the same.
CONCLUSIONS
Several new technical and conceptual advances are coming together to make this a particularly exciting time to study the chemical underpinnings of the ocean microbiome. Advances in chemical separation, spectral analyses, and data search algorithms allow for unprecedented explorations of seawater organic matter composition at the molecular level. With these powerful analytical approaches, marine organic geochemists can quickly target and identify trace components of DOM for detailed chemical characterization. A knowledge of chemical speciation greatly facilitates the interrogation of ocean metagenomic data catalogs, which can help to identify the specific microbial taxa that produce and consume organic nitrogen, phosphorus, and trace metals. Finally, using advanced microbial culture approaches, including dilution-to-extinction, marine microbiologists can quickly bring microbes with targeted metabolic capabilities into laboratory culture for detailed studies of metabolic pathways. What quickly emerges from these efforts is a picture of a complex microbiome that continually trades organic carbon, nitrogen, phosphorus, and trace metals to maximize efficient utilization and recycling of nutrients and energy. Our exploration of the marine microbiome is accelerating rapidly, but is still in its early stages. The two examples discussed in this paper both highlight how ocean chemistry, microbial genomics, and laboratory cultures can be used in concert to better understand organic phosphorus cycling and the production of methane, and the acquisition of essential iron. Both studies yielded many surprises, and as our understanding of marine microbiomes continues to evolve, we expect many new discoveries and more surprises lie ahead.
ACKNOWLEDGMENTS
We thank the Gordon and Betty Moore Foundation (Grant GBMF3298), the National Science Foundation (NSF) program in chemical oceanography (OCE-1356747), the NSF Science and Technology Center for Microbial Oceanography Research and Education (DBI-0424599), and the Simons Foundation (SCOPE Award 329108) for support of this work. R.M.B. was funded by a Linus Pauling Postdoctoral Fellowship at the Pacific Northwest National Laboratory (PNNL LDRD 69488).
REFERENCES
Boiteau, R. M., and D. J. Repeta. 2015. An extended siderophore suite from Synechococcus sp. PCC 7002 revealed by LC-ICPMS-ESIMS. Metallomics 7:877-884.
Boiteau, R. M., N. J. Hawco, D. R. Mende, M. R. McIlvin, P. N. Sedwick, M. A. Saito, E. F. Delong, and D. J. Repeta. 2016. Microbes adapt to iron scarcity through siderophore production across the eastern tropical Pacific. Proc Natl Acad Sci USA 113:14237-14242.
De Baar, H., P. Boyd, K. Coale, and M. Landry. 2005. Synthesis of iron fertilization experiments: From the Iron Age in the Age of Enlightenment. J Geophys Res 110:1-24. doi:10.1029/2004JC002601.
Gledhill, M., and K. N. Buck. 2012. The organic complexation of iron in the marine environment: A review. Front Microbiol 3:1-17. doi:10.3389/fmicb.2012.00069.
Hansell, D. A., C. A. Carlson, D. J. Repeta, and R. Schlitzer. 2009. Dissolved organic matter in the ocean. Oceanography 22:202-211.
Hansell, D. M., and C. A. Carlson (Eds.). 2015. Biogeochemistry of Marine Dissolved Organic Matter. Amsterdam: Academic Press. Chap. 2.
Karl, D. M. 2014. Microbially mediated transformations of phosphorus in the sea: New views of an old cycle. Annu Rev Mar Sci 6:279-337.
Kem, M. P., and A. Butler. 2015. Acyl peptidic siderophores: Structures, biosyntheses and post-assembly modifications. Bio-Metals 28:445-459. doi:10.1007/s10534-015-9827-y.
Mawji, E., M. Gledhill, J. A. Milton, G. A. Tarran, S. Ussher, A. Thompson, G. A. Wolff, P. J. Worsfold, and E. P. Achterberg. 2008. Hydroxamate siderophores: Occurrence and importance in the Atlantic Ocean. Environ Sci Technol 42:8675-8680.
Morel, F. M. M., and N. M. Price. 2003. The biogeochemical cycles of trace metals in the oceans. Science 300:944-947. doi:10.1126/science.1083545.
Repeta, D. J., S. Ferrón, O. A. Sosa, C. A. Johnson, L. D. Repeta, M. A. Acker, E. F. DeLong, and D. M. Karl. 2016. Marine methane paradox explained by bacterial degradation of dissolved organic matter. Nat Geosci 9:884-887.
Sandy, M., and A. Butler. 2009. Microbial iron acquisition: Marine and terrestrial siderophores. Chem Rev 109:4580-4595. doi:10.1021/cr9002787.
Sebastian, M., and J. W. Ammerman. 2009. The alkaline phosphatase Pho X is more widely distributed in marine bacteria than classical Pho A. ISME J 3:563-572.
Torres-Valdes, C. S., V. M. Roussenov, R. Sanders, S. Reynolds, X. Pan, R. Mather, A. Landolfi, G. A. Wolff, E. P. Achterberg, and R. G. Williams. 2009. Distribution of dissolved organic nutrients and their effect on export production over the Atlantic Ocean. Global Biogeochem Cycles 23. doi:10.1029/2008GB003389.
Toulza, E., A. Tagliabue, S. Blain, and G. Piganeau. 2012. Analysis of the global ocean sampling (GOS) project for trends in iron uptake by surface ocean microbes. PLoS One 7: e30931. doi:10.1371/journal.pone.0030931.
Twining, B. S., and S. B. Baines. 2013. The trace metal composition of marine phytoplankton. Annu Rev Mar Sci 5:191-215. doi:10.1146/annurev-marine-121211-172322.
Yu, X., J. R. Doroghazi, S. C. Janga, J. K. Zhang, B. Circello, B. M. Griffin, D. P. Labeda, and W. W. Metcalf. 2013. Diversity and abundance of phosphonate biosynthetic genes in nature. Proc Natl Acad Sci USA 110:20759-20764.
This page intentionally left blank.










