
7
Developing and Delivering the Next Generation of Therapies
We should be aggressive about pursuing the research and learning more and pushing ahead, because while the road may be murky, it’s there, and there’s reason for optimism and so we should just keep plugging until we get there.…
—Celia W. (Open Session Panelist)
Over the past two decades a great deal of scientific and medical research effort has resulted in progress toward developing new treatments—including potential cures—of sickle cell disease (SCD), but research is just one aspect of making novel therapies a reality. Developing the most effective therapies will require input and support from the sickle cell community, including from individuals living with the disease. This input can help guide researchers to develop new, effective treatments that are accepted by the sickle cell community. Testing new treatments in clinical trials will require a different set of considerations, including strategies for encouraging participation in trials and determining which endpoints to use as measures of success. Once novel therapies are approved by the U.S. Food and Drug Administration (FDA), considerations such as health care delivery policies and reimbursement for treatment costs will be important considerations. In each of these steps, the SCD patients’ concerns, preferences, and well-being should be given first priority.
PATIENT PERSPECTIVES
Any effort to develop a treatment or cure for SCD must start with the patient. Understanding the experiences and beliefs of individuals living with SCD, their families, and the associated community will be integral to developing effective treatments that target the most important outcomes to those stakeholders. Similarly, it is vital to understand patient perspectives concerning curative therapies to completely realize their benefits.
Although the literature in this area is lacking, researchers have identified some pervasive perspectives and attitudes within the sickle cell community and reported on the impacts of these beliefs on health outcomes. Recurring themes from this research include a distrust of the medical profession, fear of the adverse effects of therapies, inadequate SCD education and awareness, a minimization of the role of patients in shared decision making (SDM), and perceived barriers to experimental clinical studies (Bakshi et al., 2017; Haywood et al., 2011; Long et al., 2011; Scharff et al., 2010). SCD is also widely perceived to be an orphan disease that lacks sufficient resources or attention—and many believe that social factors (e.g., a majority of SCD patients are African American) explain much of this marginalized status (Bulgin et al., 2018).
Bonham et al. (2007) examined the interconnectedness of race and disease using “SCD as a prism for understanding the historical connections between genetic diseases and racial diseases, and the consequences of these connections” and viewing SCD as a foundation for “comprehending the attitudes of African Americans toward genetic research” (p. 311). Social, political, and cultural factors combined to shape the conceptualization of SCD (which has been widely seen as a “black disease”) and influence the
care given to individuals living with the disease. A study by Treadwell et al. (2006) documented general misconceptions about SCD among the African American community and also described the existence of SCD stigma and a lack of compassion and cultural sensitivity among those in the medical profession. The stigma attached to SCD, which has its roots in racism and racist attitudes, has detrimental consequences for patient health and negatively affects patient–provider relationships and care-seeking behaviors (Bulgin et al., 2018; Wailoo, 2017).
In a study examining the attitudes and beliefs of African Americans toward genetics and genetic testing, Long et al. (2011) found a lower-than-desired uptake of SCD education but reported that collaborations involving trusted sources of information, such as family members, community physicians and leaders, the church, and those with personal experiences, could help mitigate this gap. When investigating SCD patients’ perspectives on gene therapy, Strong et al. (2017) found a lack of awareness, fear and uncertainty about side effects, and concerns about the HIV-derived viral vector, infertility risks, and the potential risk for cancer. Persaud et al. (2019) investigated the perspectives of patients, parents, and clinicians toward somatic genome editing and found that patients expressed being both hopeful and fearful about the topic and also feeling insufficiently informed. The findings revealed that patients were more likely to have a positive attitude toward gene therapy when there was better education, prior participation in clinical trials, and perceived benefit. Persaud and Bonham (2018) also found that when patients have a trustful relationship with health providers, they are more likely to seek advice from providers and expect better outcomes from medical care.
Trust is a core element shaping the patient–health provider relationship, and a patient’s level of trust or distrust can affect his or her attitudes toward therapies and health outcomes. African Americans in general report higher levels of distrust of the medical community than white Americans, and sickle cell patients report concerns about biased treatment, being seen as drug seeking, and experiencing other forms of marginalization (Bonham et al., 2007; Strong et al., 2017; Wailoo, 2017). SCD patients’ trust in health teams is increased when team members are better able to relate to and communicate with them (Haywood et al., 2014; Strong et al., 2017).
A key—but often ignored—factor influencing SCD patient perspectives is how they receive information about the disease. Studies of information-seeking behaviors among African Americans have found that they rely on familial sources and other trusted sources, such as their church or peers (i.e., others living with the disease) (Acharya et al., 2009; Long et al., 2011). SCD patients report that their preferred way of receiving information about SCD is through direct interactions with medical providers,
although they are also open to other sources, such as the Internet, books and pamphlets, and DVDs, as long as the information is clear, truthful, and preferably accompanied by illustrations (Omondi et al., 2013; Strong et al., 2017). The community also recognizes the complexities of the decision-making process associated with options for disease-modifying strategies and expects to be included in that process. When deciding whether to pursue allogenic hematopoietic stem cell transplantation (HSCT), for example, patients were primarily influenced by the disease burden (particularly when it reached a critical point), consultation with HSCT clinicians, and familial support (Khemani et al., 2018). In a qualitative study of clinicians who were experts in SCD and their attitudes toward sharing decision making with patients and families regarding whether to pursue disease-modifying therapies, the clinicians were found to take a collaborative (discussing all plans) or proponent (advocating a predetermined plan) approach, depending on the disease severity and treatment urgency (Bakshi et al., 2017).
In summary, the sickle cell community has made it clear that certain factors will be key in aiding individuals with SCD to make informed decisions about curative and disease-modifying therapies, as well as effective management strategies to ensure high-quality health care. Those factors include access to trusted sources of information and resources; clear and transparent dissemination of information from the medical community, including results from clinical trials; an active and meaningful role in decision making for patients; a prioritization of patient needs; the establishment of partnerships between advocacy organizations and trusted providers; and the creation of mechanisms for building trust among providers, researchers, and the community (Lebensburger et al., 2013, 2015; Liem et al., 2010; Persaud et al., 2019). Individuals living with SCD should be empowered to advocate for themselves, must be better enabled to interact effectively with health care providers, and need an enhanced ability to navigate the health care system. Better education is needed not only for the SCD community but also for health care providers, particularly regarding patient needs, effective management, and innovative programs to ease the transition to adult care. Providers also need the training and means to address any implicit and institutional biases and other forms of systemic racism and inequities in health care.
In 2017 the American Society of Hematology (ASH) initiated an aggressive advocacy campaign related to SCD and sickle cell trait, and it set research priorities that included developing novel therapies and strengthening curative therapies. In September 2018 the National Heart, Lung, and Blood Institute of the National Institutes of Health (NIH) launched the Cure Sickle Cell Initiative to accelerate genetic therapies (NIH, 2018). These efforts bode well for the sickle cell community, as each initiative is patient-focused and committed to engaging patients, families, advocates,
and organizations in the cause. While these new developments give increased attention to curing SCD, overcoming the complicated history of race and health will be challenging.
THERAPIES UNDER DEVELOPMENT
The goal of research into new treatments is to find more effective ways to prevent or ameliorate the various manifestations of SCD—and, ultimately, to cure it altogether. The specific goals of the therapies include preventing and treating its complications, decreasing its morbidity, minimizing the numbers of crises and hospitalizations, lowering its symptom burden, prolonging life, improving quality of life (QOL), and, with a cure, eliminating SCD’s root cause. Even with an effective cure, though, ameliorative therapies will still be needed because those who have lived with the disease for years will have to address any lingering SCD-related health concerns.
As of December 2019, hydroxyurea (HU), L-glutamine, voxelotor, and crizanlizumab are disease-modifying drugs available for those with SCD, and HSCT is the only established non-experimental curative therapy (Bernaudin et al., 2007; FDA, 2019; Hsieh et al., 2009; Kutlar et al., 2019; Strouse et al., 2008). In 2017 L-glutamine became the second drug approved by FDA to prevent crises in individuals 5 years of age or older living with SCD (FDA, 2017; Riley et al., 2018), and voxelotor and crizanlizumab have been approved only recently (November 2019). None of the available therapies represent a completely satisfactory option for people affected by SCD. In particular, none of the drugs reduce the number of vasoocclusive episodes (VOEs) by more than 50 percent. However, building on decades of research, efforts are under way to develop novel pathophysiology-related agents and novel genetic approaches to a cure. This section reviews the SCD therapies, both therapeutic and curative, that are now under development.
New Drugs for SCD
In 1998, when FDA approved HU to treat acute VOEs, it was the first drug specifically for SCD—and it would remain the only such drug for nearly two decades. This was not for lack of trying, however. In the early part of the 21st century, the sickle cell community was keenly aware of the need for additional or alternate disease-modifying therapies. The adoption of HU by providers and patients alike had been associated with a number of problems, including concerns about numerous potential side effects, the need for close monitoring, the required daily long-term dosing, and concerns about long-term toxicity (Halsey and Roberts, 2003; McGann and Ware, 2015; Steinberg, 2002). Furthermore, some patients experienced
incomplete responses—or even no response—underlining the need for additional therapies (Steinberg, 1999; Stuart and Nagel, 2004). Nevertheless, drug development in SCD proceeded slowly.
Over the past two decades, however, mounting evidence from the mouse models of SCD, particularly the Berkley and Townes transgenic humanized sickle mice, has led to a more nuanced and in-depth understanding of the complex pathophysiology of SCD. The primacy of hemoglobin polymerization and red blood cell (RBC) sickling in the phenotype has not been challenged, but multiple complex and interlinked downstream mechanisms have been identified, in large part—and in some cases exclusively—based on preclinical evidence. Thus, compounding hemoglobin polymerization, cellular hyperadhesion, endothelial activation, hemolysis, hemostatic activation, oxidant stress, sterile inflammation, and hyperviscosity are now all recognized as critical determinants of the phenotype (Du et al., 2018; Kalpatthi and Novelli, 2018). This has led to a number of drugs that target specific aspects of the pathogenic cascade (see Figure 7-1).
These drugs, along with their novel therapeutic strategies, are typically classified into six categories, as summarized in the table in
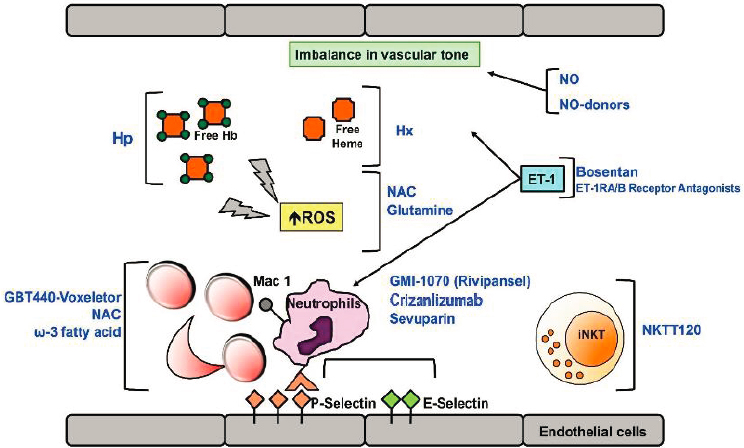
NOTE: Ab = antibody; ET-1 = endothelin-1; ET-R = endothelin-1 receptor; Hb = hemoglobin; Hp = haptoglobin; Hx = hemopexin; iNKT = invariant natural killer T cells; NAC = N-Acetyl-cysteine; NKTT120 = humanized monoclonal antibody specifically depleting iNKT; NO = nitric oxide; ROS = reactive oxygen species.
SOURCE: Matte et al., 2019.
Appendix I: anti-sickling agents, anti-adhesion agents, antioxidant agents, anti-inflammatory agents, anticoagulant and antiplatelet agents, and nitric-oxide (NO)-related agents. All of these treatments share certain limitations: they do not address the genetic cause (the hemoglobin S [HbS] mutation), they each address only certain manifestations of the disease, they do not specifically target chronic pain, and they cannot reverse end-organ damage. Nonetheless, they hold promise for interrupting the pathophysiologic mechanisms in SCD and thus decreasing organ-specific complications, reducing morbidity and mortality, and improving QOL. More details can be found in Ballas et al. (2012), which provides an extensive review of SCD management based on the complications and notes the variation among patients and in the same patient over time. Other reviews discuss advances in treatment strategies targeting inflammation, oxidative injury, vascular tone, hemoglobin polymerization, and adhesion (Ataga and Desai, 2018; Kapoor et al., 2018; Steinberg and Brugnara, 2003; Telen et al., 2019).
Agents That Block HbS Polymerization
HbS polymerizes in RBCs to create fibers that reduce the flexibility of RBCs leading to hemolysis and other downstream complications, so one approach to treating SCD is to block HbS polymerization. There are various approaches to doing this. One is to increase fetal hemoglobin (HbF) production. High intracellular HbF levels prevent or reduce HbS polymerization, as evidenced by individuals with congenital hereditary persistence of HbF co-inherited with SCD, although the protection from sickling is usually not complete. In addition to HU, whose role in therapy is well established and has been confirmed by strong evidence obtained in the United States (Charache et al., 1995; Steinberg et al., 2010) and throughout the world (Tshilolo et al., 2019; Voskaridou et al., 2010), other drugs, such as decitabine (Molokie et al., 2017), histone deacetylase inhibitors (Shearstone et al., 2016), sodium 2,2-dimethylbutyrate (Kutlar et al., 2013), and metformin (Han et al., 2019), have been found to boost HbF levels and are under investigation for SCD.
Another strategy to reduce HbS polymerization involves modulating the oxygen affinity of hemoglobin. Voxelotor (GBT440), which was approved by FDA for SCD in November 2019, increases the oxygen affinity of hemoglobin (Vichinsky et al., 2019). By increasing the oxygen affinity of hemoglobin, voxelotor maintains hemoglobin in the oxygenated state and reduces polymerization and sickling (Vichinsky et al., 2019). Clinical trials in adults and children have shown reduced hemolysis and improved hemoglobin, although these changes were not accompanied by a statistically significant reduction in VOEs (Vichinsky et al., 2019). While voxelotor does not appear to adversely affect oxygen delivery, according to measurements
of erythropoietin levels in study patients, more research may be needed in children at a high risk of stroke, in whom cerebral oxygenation partly relies on increased oxygen extraction. There are also lingering concerns that increasing the total hemoglobin without reducing the intracellular concentration of HbS may lead to hyperviscosity and its attendant complications (Estepp, 2018).
Anti-Adhesion Agents
Cellular hyperadhesion is a critical component of vaso-occlusion. In the proinflammatory milieu of SCD, the endothelium and the blood cells overexpress activated adhesion molecules (integrins), which promote the binding of sickle RBCs, reticulocytes, leukocytes, and platelets to the endothelium and to one another. The complex network of interactions between cellular elements and the endothelium is amply documented in animal models (Sundd et al., 2019). The translational relevance of paradigms developed from mouse research has now been validated in humans. Among the most promising approaches are those that target selectins. P-selectin, E-selectin, and several integrins mediate cellular adhesions in mice (reviewed in Telen, 2007). FDA approved crizanlizumab for SCD in November 2019 (FDA, 2019). Crizanlizumab, a monoclonal antibody that targets P-selectins, has been tested in humans and found to prevent VOEs, as predicted by the evidence in mouse models. High-dose crizanlizumab reduced the rate of VOEs to a median of 1.63/year compared with 2.98/year with placebo (a 45.3 percent lower rate, p = 0.01) (Ataga et al., 2017; Kutlar et al., 2019). In addition, “the median time to the first crisis was significantly longer with high-dose crizanlizumab than with placebo (4.07 versus 1.38 months, p = 0.001), as was the median time to the second crisis (10.32 versus 5.09 months, p = 0.02)” (Ataga et al., 2017, p. 429). Rivipansel, unlike crizanlizumab, is a pan-selectin inhibitor with particular activity against E-selectin (Chang et al., 2010). Rivipansel has shown clinical benefit in a Phase II trial, where it reduced cumulative intravenous opiate requirements by 83 percent in patients hospitalized with VOEs (Telen et al., 2015); however, in a major setback for the field, Pfizer recently announced that the Phase III trial (RESET) did not meet the primary endpoint of time to readiness for discharge and the key secondary efficacy endpoints of time-to-discharge, cumulative IV opioid consumption, and time to discontinuation of IV opioids (Pfizer, 2019). The reason for the negative trial results may lie in either the inferiority of E-selectin blockade (as compared to P-selectin blockade) as a strategy to block VOEs or in the difficulty of aborting a VOE once it has started. If the latter hypothesis is correct, preventive approaches will remain more successful. A third selectin inhibitor, sevuparin, which has predominant activity against L-selectin, is also under investigation (White et al.,
2019). While side effects in the anti-selectin trials were relatively modest, heightened surveillance for hemorrhagic and infectious complications will be warranted in clinical use, given the role of selectins in hemostasis and immunity. Finally, intravenous immunoglobulins are under investigation for their capacity to block neutrophil adhesion to RBCs and the endothelium.
Antioxidant Agents
Oxidant stress is the by-product of multiple disturbed pathways in SCD. Both reactive oxygen and nitrogen species are elevated and natural antioxidant mechanisms are depleted in the plasma and tissues. Enzymatic sources of reactive oxygen species (ROS), including xanthine oxidase (Aslan et al., 2001), nicotinamide adenine dinucleotide phosphate oxidase (George et al., 2013), and NO synthase (Kaul et al., 2000), are upregulated in SCD and promote oxidant damage in response to ischemia-reperfusion injury. Autoxidation of RBCs and platelet mitochondrial ROS production (Cardenes et al., 2014) are also sources of oxidant stress. Finally, hemolysis leads to free hemoglobin quenching NO in the vasculature by the Fenton reaction (Minneci et al., 2005). Restoring the balance of oxidants and antioxidants is a viable therapeutic strategy in SCD that is finally bearing fruit. L-glutamine is an antioxidant and precursor of NO that was approved as the second disease-modifying drug (Niihara et al., 2018). While its benefit is more modest than that obtained with HU (25 percent versus 50 percent reduction in VOEs), L-glutamine has a much more favorable side-effect profile (Charache et al., 1995; Niihara et al., 2018). Omega-3 fatty acids also have antioxidant and anti-inflammatory properties and have been shown to reduce VOE frequency by approximately 50 percent (Tomer et al., 2001); they are currently being investigated in larger, randomized studies.
Anti-Inflammatory Agents
A chronic state of sterile inflammation is a cardinal feature of SCD. Its role is most evident in acute chest syndrome (ACS), where inflammation triggers capillary leak and acute lung injury (Sundd et al., 2019), but the hallmarks of inflammation are present at steady state, where levels of inflammatory cytokines, leukocytes, and other pro-inflammatory molecules are elevated as compared with individuals without SCD (Schimmel et al., 2013). By-products of hemolysis (i.e., hemin) and ischemia-reperfusion injury are responsible for activating vascular cells and producing damage-associated molecular products and proinflammatory cytokines (Chen et al., 2014; Dutra et al., 2014). Intensive research in mice has identified multiple pathways of inflammation, including hemin-TLR4, multiple mediators
of the inflammasome complex, and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)1 family of transcriptional factors, leukotrienes, and mast cells (Belcher et al., 2014; Kaul and Hebbel, 2000). There are many compounds under development that target specific aspects of inflammation. Corticosteroids could potentially target multiple pathways, but their role in SCD remains controversial after clinical evidence of rebound pain and VOE emerged following their use in severe ACS (Griffin et al., 1994). Clinical trials are ongoing with efforts to modulate specific mediators of inflammation, which could potentially lead to drugs with more limited and predictable side-effect profiles. The drugs under investigation include leukotriene inhibitors (e.g., zileuton and montelukast), inhibitors of mast cell activation (e.g., imatinib and cromolyn sodium), inhibitors of natural killer cells (e.g., rogadenoson and adenosine A2A receptor activators), inhibitors of NF-κB (e.g., sulfasalazine), and components of the inflammasome complex (e.g., canakinumab and NLRP3 inhibitors) (Field et al., 2017; Telen, 2016; Vincent et al., 2013).
Anticoagulant and Antiplatelet Agents
Hemostatic activation is present at baseline in SCD and worsens during VOEs. Virtually all components of the coagulation cascade are affected in SCD (Ataga and Key, 2007; Ataga et al., 2008; De Franceschi et al., 2011; Peters et al., 1994; Setty et al., 2008; Singer and Ataga, 2008; Stuart and Setty, 2001; Whelihan et al., 2016). Externalization of RBC prothrombotic phospholipids (Franck et al., 1985), RBC and platelet microparticle shedding (Allan et al., 1982; Wun et al., 1998), hemolysis-mediated platelet activation (Cardenes et al., 2014; Villagra et al., 2007), and the reduced clearance of prothrombotic cells post-splenectomy (Crary and Buchanan, 2009) are all documented mechanisms of hemostatic activation in SCD. Clinical studies in large administrative databases, smaller case series, and single-institution reports have shown a higher prevalence and incidence of pulmonary thrombosis and intravascular catheter-related thrombosis in SCD, particularly in hospitalized patients (Brunson et al., 2017, 2019; Naik et al., 2013, 2014; Novelli et al., 2012; Stein et al., 2006). Yet, the association between hemostatic activation and SCD complications is less clear. Theoretically, antiplatelets and anticoagulants should reduce vaso-occlusion by dampening hyperadhesion and platelet recruitment. However, clinical studies of anticoagulants and antiplatelet agents have largely been disappointing (Noubouossie et al., 2016). There is ongoing research on recently approved drugs, including the newer direct oral anticoagulants,
___________________
1 NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a protein complex that controls transcription of DNA, cytokine production, and cell survival.
which are currently being investigated in clinical trials for their potential to mitigate the SCD phenotype. It remains to be determined whether antiplatelet agents may be beneficial in subsets of patients with a more pronounced hemostatic activation profile and as a part of multidrug interventions. For instance, in the Determining Effects of Platelet Inhibition on Vaso-occlusive Events clinical trial of prasugrel, an agent that inhibits platelet activation, a subset analysis of the pediatric study population showed a possible benefit in the adolescent group (12–17 years old) (Heeney et al., 2016).
Nitric Oxide
NO metabolism and metabolites are profoundly altered in SCD and central to its pathophysiology. NO therapeutics, including inhaled NO, L-arginine, and sildenafil (a phosphodiesterase inhibitor), have been tested in clinical trials. The results have been mixed, with the DeNOVO trial being unquestionably negative (Gladwin et al., 2011) and others inconclusive or terminated early due to toxicity (Walk-PHaSST) (Machado et al., 2011) or low enrollment (Children’s Hospital Los Angeles and Hope Pharmaceuticals, 2009). Yet, NO modulation remains an attractive therapeutic strategy, particularly for specific complications or in the subset of patients with particularly brisk hemolysis. Two potential treatments under investigation are a topical nitrite preparation for leg ulcers, and the oral soluble guanylate cyclase stimulator riociguat for patients with a hyperhemolysis phenotype (STERIO-SCD trial) (Kato, 2015). There is also preliminary evidence that L-arginine, a NO precursor, may reverse platelet mitochondrial ROS production and thereby reduce platelet activation and other downstream effects (e.g., hyperadhesion). L-arginine reduced the frequency of VOEs in children (Morris et al., 2013) and is under investigation in a larger study.
Defining a Cure
As noted above, neither the drugs already approved by FDA for use in SCD nor any of the drugs under development address the root cause. They focus instead on addressing the manifestations of the disease. Thus, much attention has been paid to developing a “cure” that would eliminate the underlying mutation and shut down the pathways that lead to the various manifestations. Ideally, a cure would permanently correct the mutation and thus eliminate VOEs, arrest progressive organ damage, and possibly even reverse some pre-existing organ damage. However, even such a therapy would not completely “cure” those whose bodies have been damaged by SCD; they would likely still have organ damage, experience chronic pain, and continue to require treatments.
There are currently two basic approaches to curing SCD, one available now and another, seen as more promising, that is still under development. The first is HSCT. It is the only curative therapy available, but it has shortcomings that limit its value, including difficulties finding suitable donors, the possibility of the transplant being rejected, and short-term morbidity and mortality risks (Gluckman et al., 2017; Hsieh et al., 2014; Thompson, 2013; Walters, 2015). The second is the “holy grail” of gene therapy, or gene editing, which does not rely on donors and circumvents the challenges of graft-versus-host-disease (GVHD) and the need for immunosuppression past transplantation (Walters, 2005). It is still in the experimental stages, however, and is not widely available; it has its own set of challenges; and it has been met with a somewhat less than enthusiastic response from the SCD community because of insufficient education and a fear of known and unknown risks (Persaud et al., 2019; Strong et al., 2017).
Stem Cell Transplant
Since the 1990s, stem cell transplant has offered the potential for an SCD cure. More than 1,000 patients, predominantly in the United States and Europe, have now been treated with allogeneic transplantation with excellent results (Gluckman et al., 2017; Hsieh et al., 2014; Walters et al., 1996, 2001). The finding that even modest levels of donor chimerism (i.e., 20 percent myeloid engraftment) result in significant levels of donor hemoglobin that prevent HbS polymerization has led to the development of non-myeloablative or reduced-intensity conditioning regimens; the cumulative outcomes of both remain encouraging. The outcome of transplants from matched siblings has been particularly impressive. The European Blood and Marrow Transplant, Eurocord, and the Center for International Blood and Marrow Transplant Research reported the results on 1,000 recipients of human leukocyte antigen (HLA)-identical sibling transplants performed between 1986 and 2013 and found a 5-year event-free and overall survival of 91.4 percent (95% confidence interval [CI] 89.6–93.3) and 92.9 percent (95% CI 91.1–94.6), respectively (Gluckman et al., 2017). Event-free survival was lower with increasing age at transplantation (hazard ratio [HR] = 1.09; p < 0.001) and higher for transplantations performed after 2006 (HR = 0.95, p = 0.013) (Gluckman et al., 2017). Increasing age was associated with higher rates of acute and chronic GVHD (4 percent and 2 percent higher HR for every 1 year of age increment) and graft failure or death (10 percent higher HR for each year) (Gluckman et al., 2017). The higher risk of chronic GVHD and need for fertility preservation may be indications for non-myeloablative conditioning in older patients (Bernaudin et al., 2019).
Patients who have stable donor engraftment are cured and no longer experience VOEs and other acute complications. However, chronic pain
lingers in those who were significantly affected by it before the transplant, and it is unclear whether and to what extent the organ damage can be reversed by transplantation. Stem cell transplantation has largely remained limited to patients with matched siblings, performed outside of clinical trials, and out of reach for most adults with organ dysfunction. Long-term toxicity and reproductive risks also remain a concern. Newer research avenues aim to develop safer conditioning regimens, effective GVHD prevention strategies, more widely available means of fertility preservation, and to expand access to those without matched siblings.
Unrelated-donor stem cell sources are actively sought to expand access to transplantation in SCD and other benign and malignant hematological conditions. While the probability of identifying fully matched donors in the bone marrow registry is low for African Americans (19 percent), allowing for a partial mismatch at one of eight HLA loci increases the likelihood of finding a donor to more than 70 percent (Gragert et al., 2014). Immunologic mismatch leads, however, to a higher incidence of GVHD. While this may be a tolerable side effect in transplant recipients with hematologic malignancies due to the concurrent graft-versus-leukemia effect, it is highly undesirable in patients with benign hematologic conditions, who do not derive any appreciable benefit from GVHD. A recent report shows that when a sibling donor is not available, the outcome of transplantation from different sources is similar. Event-free survival between recipients of transplants from haploidentical related donors (HR = 1.43, 95% CI 0.81–2.50; p = 0.21) or mismatched unrelated donors (HR = 1.17, 95% CI 0.67–2.05; p = 0.58) versus HLA-matched unrelated donors, or mismatched unrelated donors versus haplo-identical related donors (HR = 1.22, 95% CI 0.65–2.27; p = 0.98), was not statistically significantly different (Eapen et al., 2019). Umbilical cord blood has been explored as a stem cell source due to its lower risk of inducing GVHD (Bernaudin et al., 2007), but its longer time to engraftment and the resulting delayed hematological and immune recovery and low graft cell volume have yielded high graft rejection rates (Kamani et al., 2012; Saraf et al., 2016). Techniques to expand the umbilical cord graft volume ex vivo are under development (Horwitz et al., 2014).
Side Effects and Risks
The side effects and risks of allogeneic stem cell transplant are similar to those observed in other diseases, but they are compounded by SCD-specific factors. For instance, the toxicity of conditioning regimens and immunosuppressive agents after transplantation may be poorly tolerated by organs (e.g., kidneys or organs in the cardiovascular system) whose physiologic functional reserve is depleted by SCD. The immune reactivity of the transplant recipient is also affected by chronic transfusion regimens, which may result
in HLA and RBC alloimmunization and alterations in the bone marrow microenvironment from marrow infarction and stress erythropoiesis. There are recent guidelines for the screening and early recognition of complications after transplantation for hemoglobinopathies (Shenoy et al., 2018).
Stem cell transplant in SCD also poses unusual ethical concerns. Unlike hematologic malignancies, which are often rapidly lethal without curative transplantation, SCD is a chronic disease, and certain subgroups discussed in Chapter 1, such as those with HbSC, HbS/beta thalassemia, or HbSS with hereditary persistence of fetal hemoglobin, have life expectancies that may be comparable to those of unaffected African Americans (Platt et al., 1994). In other patients, existing and developing disease-modifying strategies offer the promise of long-term survival with tolerable morbidity and acceptable QOL. Thus, the side effects of stem cell transplantation need to be assessed in the context of the patient’s individual experience with SCD and projected disease course. With matched sibling transplantation, the side effects are relatively modest, and long-term, disease-free survival is the norm. However, as the immunological mismatch becomes more pronounced, the risks of severe toxicity, such as GVHD, rise dramatically, particularly in adults. Opportunistic infections caused by the immunosuppressive regimens also remain a concern although they have been mitigated by non-myeloablative and reduced intensity conditioning regimens.
Stem cell transplant has profound psychosocial repercussions from pre-transplant through the post-transplant phase that must be prevented or managed. An assessment of the psychological well-being of the transplant recipient should be conducted early on, and pre-/post-transplant assessments should include evaluations by social workers and other mental health professions. It is critical to carefully address the family and socioeconomic support available to the transplant candidate. Caregivers’ perspectives may weigh significantly in decisions and should be solicited (Khemani et al., 2018). Thus, an SDM model that includes that patient’s support system should be adopted throughout the process of informed consent and the treatment and follow-up period. Potential barriers to adherence to anti-rejection regimens should be addressed. It is unrealistic to expect that most patients on long-term opiates will be able to transition to non-opiate analgesia in the immediate post-transplant period. First, certain painful complications, such as avascular necrosis, are not reversible, nor is neuropathic pain. Second, central sensitization and the interplay among decreased executive function, mood disorders, and pain are also expected to have long-term repercussions. Thus, opiate therapy before transplant should be streamlined, and an effort should be made to ease transition by exploring non-pharmacological analgesic strategies, optimizing mental health, and addressing spiritual and existential concerns in advance. Even when stem cell transplant is relatively uneventful and a cure is attained, there should be
ongoing attention to the psychological needs of the recipients. As a chronic, lifelong, life-threatening illness, SCD shapes self-identity and psychological ownership. In other words, it may become part of the patient’s “identity” (Karnilowicz, 2011), which can then be questioned or threatened by a cure.
Implications for Fertility and Other Reproductive Issues
Gonadotoxic conditioning regimens with busulfan, other myeloablative drugs (e.g., cyclophosphamide), and radiation affect fertility and carry a risk of ovarian failure (prevalence of 65–84 percent) (Joshi et al., 2014; Loren et al., 2011), particularly in post-pubertal individuals. Furthermore, transfusional hemosiderosis arising from repeated transfusions in the pre-transplantation period also predisposes to endocrinopathy and reduced fertility. Thus, fertility preservation should be discussed up front and offered by means of ovarian and testicular tissue cryopreservation whenever possible and as recommended by the Practice Committee of the American Society for Reproductive Medicine 2013 guidelines (ASRM, 2013), with the caveat that it may not be covered by health insurers outside of a clinical trial. In European countries where the procedure is at no cost to the patient, such preservation is performed systematically in all SCD transplant patients (Bernaudin et al., 2019). However, both laparoscopic unilateral oophorectomy and testicular tissue explant are invasive and require general anesthesia, which predisposes patients to vaso-occlusive complications in the post-operative period. Thus, adequate precautions to minimize post-procedure complications should be enacted. For settings where pre-transplantation sperm and ovarian cryopreservation are unavailable or unaffordable, fertility-preserving conditioning regimens are being developed, and gonadal shielding during irradiation is an option.
Another reproductive concern related to stem cell transplant is the need for subsequent hormonal therapy to develop secondary sexual characteristics in prepubertal children, particularly girls (Dallas et al., 2013; Walters et al., 2000). It is important to frame the discussion of the reproductive risks of stem cell transplantation (Xue et al., 2019) within the broader context of the risks deriving from untreated SCD, which is known to affect sexual maturation and fertility, and alternative treatments that may also affect fertility, such as HU (Joseph et al., 2019; Pecker et al., 2019).
Expanding Access to Non-Matched Donors and Others
Access to stem cell transplant continues to be very limited. Only 14 percent of individuals with SCD will have an HLA-matched sibling donor (Walters, 2015). Similarly, only 19 percent will have a well-matched unrelated donor, far lower than the 75 percent likelihood for Caucasian patients
(Walters, 2015). In contrast, it is estimated that more than 50 percent of patients will have a haplo-identical donor. Haplo-identical transplant for nonmalignant diseases has expanded exponentially over the past few years thanks to new conditioning strategies, such as the addition of thyotepa to the preparative regimen (de la Fuente et al., 2019), graft preparation by T-cell depletion and CD34+ selection to reduce GVHD, and post-transplant cyclophosphamide to modulated alloreactivity (Brodsky et al., 2008). While relatively few patients with SCD have received a haplo-identical graft, the results have been encouraging. The rates of GVHD and graft rejection are lower than initially predicted (although long-term follow-up data are not yet available), and the majority of patients have experienced resolution of their anemia and other SCD-related complications (Cairo et al., 2019). Several clinical trials of haplo-identical transplantation with improved conditioning regimens or posttransplant immunosuppression are under way (Limerick and Fitzhugh, 2019; Tanhehco and Bhatia, 2019).
Long-Term Outcomes
Studies of the long-term outcomes after transplantation have explored the potential to stop disease progression or reverse the most severe complications. There is emerging evidence that both neurological and cardiovascular complications may either remain stable or improve. Secondary strokes have not occurred after transplantation (Bernaudin et al., 2007; Walters et al., 2010), and overall cerebral vasculopathy, including silent cerebral infarcts and lacunar infarcts, has remained stable, as recorded by magnetic resonance imaging (MRI) (Green et al., 2017), except in those patients with the highest cerebrovascular disease burden (Dallas et al., 2013). Transplant-related neurological toxicity, such as posterior reversible leukoencephalopathy syndrome, may sometimes complicate the assessment of neurological outcomes after transplantation.
Pulmonary hypertension is a major risk factor for early mortality, with a tricuspid regurgitant jet velocity > 2.5 m/s conferring a relative risk (RR) of 10 for mortality (Gladwin et al., 2004). After transplantation, tricuspid regurgitant jet velocity improved from 2.84 m/s (95% CI 2.71–2.99) before HSCT to 2.33 (95% CI 2.14–2.51) over 3 years in adults (Hsieh et al., 2014). Pulmonary function tests also showed stable deficits or improved biomarkers (Gilman et al., 2017; Walters et al., 2010).
A major caveat concerning studies of long-term transplant outcomes is that a control group receiving best supportive care, transfusions, or HU has not generally been available for comparison.
Research Gap: Outcomes for Pediatric Patients Versus Adults
The optimal timing of transplantation in children with an identical HLA-matched sibling remains unknown, but mounting research evidence suggests that outcomes are superior in patients younger than 10 years old. There is also a strong rationale to carry out transplants in patients before complications and irreversible organ damage have occurred. In both children and adults, common indications for transplantation have included a severe phenotype despite HU or transfusion therapy or any severe complication, such as pulmonary hypertension or progressive cerebral vasculopathy (Kassim and Sharma, 2017; Walters et al., 1996). Yet criteria to recommend transplantation mostly rest on expert opinion, without any validated tools to determine optimal timing or suitability. Biomarkers to predict disease course, major outcomes, and mortality are still needed (Kalpatthi and Novelli, 2018).
Research is needed on patients’ QOL after transplant, particularly in adults; extensive studies documenting QOL pre- and post-transplant are not yet available. Algorithms and decision trees based on biomarkers of disease severity are also not available and are urgently needed for use in selecting candidates for transplantation. Ideally, transplantation should be prioritized in those patients with a severe phenotype despite disease-modifying therapies but whose organ function remains adequate to withstand the physiologic demands of the procedure. Decisions concerning transplantation opportunity and timing will become even more complex once the new biological therapies under development become available. A future is foreseeable where multiple medications used sequentially or in combination may significantly mitigate the phenotype to the point that stem cell transplantation offers no added value. Funding for biomarker development is imperative for generating the information needed to navigate these complex scenarios.
Another important area of research concerns whether transplantation can reverse specific complications. In particular, it will be important to ascertain whether the progressive deterioration in neurocognitive function in SCD can be slowed or reversed.
Research on chronic pain and its effects on mood in SCD is urgently needed and will be important in dissecting the multifaceted pain that patients often continue to experience after transplantation. In general, research on the psychological impact of transplantation will be needed, particularly in the arena of self-identity and perceptions of the self in relation to others.
Finally, more studies are needed to analyze the patients’ and caregivers’ perspectives on important factors that affect decision making in transplantation (Khemani et al., 2018).
Gene Therapy
Gene therapy was an unattainable goal for SCD until the past decade. However, successes with primary immunodeficiency syndromes and hemophilia have spurred interest in gene therapy approaches for hemoglobinopathies. Since the 1990s, the use of viral vectors has allowed ex vivo gene therapy (via the insertion of genes into autologous hematopoietic stem cells). Most recently, the development of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) technology has offered the promise of a cure (Jinek et al., 2012). Both approaches are preferable to stem cell transplant because they overcome the problem of a lack of suitable donors and also the risk of transplant-related complications, such as GVHD and opportunistic infections from prolonged immunosuppression. The principal methods of gene therapy under development are (1) the addition of b-globin or bT87Q-globin to produce hemoglobin (HbA) or γ-globin to enhance HbF levels, (2) HbF induction by editing of globin regulatory elements or knockdown of HbF repressors, and (3) direct gene correction of the SCD mutation with programmable nucleases (Demirci et al., 2019).
Gene Replacement
The introduction of replication-defective, HIV-1-based lentiviral vectors (LVs) has overcome many of the limitations of older gammaretroviral vectors, including the inability to transduce quiescent hematopoietic stem cells and to carry large gene constructs, such as b-globin and its regulatory elements. Genetic transfer of an anti-sickling b-globin LV into hematopoietic stem cells followed by myeloablative transplant cured one child (Ribeil et al., 2017), but it was not successful in seven subsequent adults with sickle cell anemia, which led to modifying the intensity of the conditioning regimen and improving the stem cell dose and gene transfer protocol (Kanter, 2017). Promising preliminary reports have been presented at the ASH annual meeting in 2018, in 2019 at the American Society of Gene and Cell Therapy meeting, and at European Hematology Association meetings. One study is a Phase I/II trial of modified gamma globin LV-based gene therapy in two adults with SCD after a reduced-intensity conditioning regimen. In this trial, the expression of a modified HbF (HbFG16D) was shown to be 20 percent for the first patient 1 year post-transplant, with a similar trajectory for the second patient, who was still in the early phase of post-transplant at the time of publication of the results; these findings were associated with a reduction in acute pain episodes (Malik et al., 2018). Another study, a Phase I trial of lentiviral-based gene transduction of a b-globin with an anti-sickling substitution
(T87Q) into bone marrow harvested or plerixafor-mobilized stem cells in 15 patients (Tisdale et al., 2018) showed more robust production of HbAT87Q (Kanter et al., 2019; Mpara et al., 2019). In four patients with more than 6 months follow-up, HbAT87Q was 47–60 percent of total hemoglobin, almost equaling or exceeding HbS levels (Kanter et al., 2019), and it was accompanied by an improvement in the SCD phenotype. Based on up to 3 years of follow-up, insertional mutagenesis and oncogenesis, also major concerns with gammaretroviral vectors, have not materialized in patients with thalassemia or SCD treated with lentiviral-based gene therapy, but longer follow-up is required.
Given the variability of results with the ongoing gene therapy approaches, stopping points for trials need to be defined and criteria developed to prevent the possibility that noncurative studies (where the gene product is not likely to be expressed to a level necessary to achieve a cure) are misclassified as curative.
Gene Editing
CRISPR/Cas9 has revolutionized gene editing by providing an efficient, easy-to-design approach that is less costly than those that rely on nucleases. Gene-editing strategies aim at suppressing HbS polymerization via the following major mechanisms: (1) inducing HbF by inhibiting the binding of transcriptional repressors BAF chromatin remodeling complex subunit BCL11A (BCL11A) and leukemia/lymphoma-related factor, or targeting transcriptional regulators of HbF, (2) correcting the HbS mutation, or (3) inserting an anti-sickling beta-globin cDNA (betaAS3). Approaches are being developed to target hematopoietic stem cells and inducible pluripotent stem cells. Concerns exist about the potential immunogenicity of guide RNAs or Cas9 and the efficiency and specificity of editing. The most advanced gene editing approach involves deleting BCL11A; it is part of an ongoing Phase I/II clinical trial by CRISPR Therapeutics and Vertex Pharmaceuticals. In October 2019, CRISPR appeared to be successful in restoring the functional bone marrow cells in the first patient with SCD to undergo this gene editing technique (Stein, 2019).
Ethical Issues and Other Considerations
Gene therapy shares some of the concerns surrounding stem cell transplantation because it requires a conditioning regimen, with the attendant risks of infertility and secondary malignancies, particularly when myeloablative regimens are employed, as in ongoing gene therapy approaches. In gene therapy, as in allogeneic transplantation, there are also uncertainties as to the optimal source of stem cells. Mobilizing autologous peripheral
blood stem cells in patients with SCD is now feasible thanks to plerixafor, a molecule that releases stem cells from their marrow niches into the circulation and, unlike filgrastim, does not appear to cause side effects (Boulad et al., 2018; Esrick et al., 2018; Hsieh and Tisdale, 2018; Lagresle-Peyrou et al., 2018). It remains to be determined whether peripheral blood or bone marrow is the optimal source of autologous stem cells for gene transfer or editing. Maximizing the stem cell yield is critical, because many gene transfer approaches require culturing stem cells ex vivo, which may lead to a loss of repopulating potential and ultimately an inadequate number of cells harboring the transgene for reinfusion.
A well-publicized ethical concern surrounding all gene therapy approaches is their potential to be adapted to germ line editing (in human embryos). NIH and many regulatory agencies throughout the world do not support this application of the technologies (Evitt et al., 2015). There are additional ethical considerations about safety, particularly surrounding immunogenicity in CRISPR/Cas9 and the lingering concerns about mutagenesis, given the relatively short duration of the follow-up of the treated patients to date. When devising therapies for a disease that is disproportionally common in low- to middle-income areas of the world, it also behooves investigators and policy makers to promote widely applicable approaches that do not rely on highly specialized infrastructure and expertise. Thus, approaches should also be developed that target stem cells in vivo, a less technically challenging procedure.
Finally, similar to the situation with stem cell transplantation, the SCD community has lower risk tolerance for gene therapy due to the availability of effective alternative therapies and the absence of long-term outcomes data for most patients. Thus, it is paramount that gene therapy trials receive the appropriate oversight by both an FDA-appointed and a non-FDA panel of experts. There also needs to be greater assurance for postmarket evaluation and controlled use because in rare diseases, such as SCD, widespread use of novel disease modifiers by providers not familiar with the disease can lead to dangerous and devastating outcomes. The analysis of long-term data and expert guided therapy will be crucial.
The technical limitations of current gene therapy approaches need to be underscored. First, all current strategies involve ex vivo gene transduction or editing, which involve the ex vivo manipulation of stem cells, with their potential loss of repopulating potential, and myeloablative chemotherapy to accommodate the auto transplant. Ideally, both problems will be overcome by in vivo gene therapy approaches, where the viral vector will be directly injected into the patient, as it is the case in techniques that are under investigation for hemophilia (Nienhuis et al., 2017). Second, it is important to note that gene therapy does not necessarily equate to a cure (i.e., the permanent and complete suppression of HbS polymerization and its
attendant complications) and should never be labeled and presented as such to the patients and other stakeholders. Based on the preliminary results of the ongoing gene therapy trials, it is unlikely that any of the current gene therapy approaches will result in a cure, although it is hoped that progressive technical advances will eventually achieve a cure in the next decades.
Family Privacy Concerns
Any procedure that accesses DNA poses ethical concerns—related to privacy, confidentiality, subsequent use, and disclosure to significant others—that have legal and societal repercussions (NASEM, 2017a).
CLINICAL TRIALS AND THE DRUG APPROVAL PROCESS
The final step in the drug development process is the clinical trials necessary to receive approval from FDA to make the drug widely available, and the drugs in development can face some special challenges arising from SCD characteristics and affected individuals’ circumstances. One set of challenges is related to the African American community’s experiences with the U.S. health care system and the resulting lack of trust that many members have for doctors and the entire health care system. A second set of challenges stems from the multiplicity and complexity of SCD symptoms, which makes it difficult to determine the best way to measure a treatment’s efficacy.
Patient Mistrust and Lack of Awareness
Clinical trials require an awareness of the existence of the trials and the willing participation of individuals with the disease, which in turn requires a certain level of trust among those individuals. Addressing these issues will help smooth the way for successful clinical trials of the SCD treatments now in development. Because individuals of African descent are significantly more likely to have the SCD mutation than people whose ancestors come from other parts of the world, U.S. clinical trials of SCD drugs will inevitably require large percentages of African Americans among the trial participants, but African Americans have many reasons to distrust the health care system.
First, the history of human experimentation in the United States has been marked by racism and inequality, as epitomized by the infamous U.S. Public Health Service Syphilis Study (CDC, 2015). Furthermore, many African Americans have experienced institutionalized racism in health care and the research community. Patients with SCD, specifically, have
suffered from the stigma of an inherited disease that causes intractable pain (Blake et al., 2018; Bulgin et al., 2018); because that pain is almost never “objectively” documented, legitimate requests for opiates have often been misconstrued as “drug seeking” behavior, particularly in emergency departments (EDs) (Shapiro et al., 1997). These misperceptions among health care providers of addiction and abuse are now being compounded by a regulatory and institutional environment that curtails opiate prescribing to control the U.S. opiate crisis (NASEM, 2017b), with SCD patients at risk of experiencing unintended effects in this climate. Regular misunderstandings between SCD patients and health care providers over pain control have often generated mutual mistrust (Puri et al., 2016). None of this is conducive to a successful partnership between patients and providers to test novel SCD treatments.
Compounding the issue of mistrust, decreased access to care among African Americans and a lack of clinicians specializing in caring for patients with SCD have resulted in a lack of awareness of new therapies and clinical trials among SCD patients. In response, ASH has recently developed several initiatives to increase access to care, educate providers across the nation, and bolster research infrastructure, including developing a clinical trial network (Michaelis, 2019).
Social media is emerging as a powerful tool with which to disseminate health care information and help shape opinions. For instance, two Facebook groups, the Sickle Cell Warriors and Sickle Cell Unite, boast thousands of users and have become important resources for the community through the information and support they provide to members and users. The discussions in these groups can also provide insight into perceptions among stakeholders, such as their opinions on the efficacy of HU. One published analysis of common themes among Facebook group members reported that some patients and caregivers perceived HU as masking symptoms (e.g., by artificially improving blood counts), thereby making it more difficult for patients to receive necessary acute care for pain, while others thought of HU as a “cancer drug” (its original approved use); both perceptions likely make patients less willing to take the drug (Walker et al., 2019). One implication of this research is that social media could be leveraged by health care providers to advance sickle cell research agendas and dispel misconceptions about clinical trials.
Clinical Evidence for Approval
Clinical trials require an objective way to measure the outcomes achieved with the tested therapy and compare those outcomes with those that were achieved with other therapies or no therapy. However, this is not always a straightforward process for SCD.
Determining Which Endpoints to Use
A key question that must be answered before running a clinical trial is which endpoints will be used to judge the outcome. With SCD, however, developing biomarkers to use as endpoints in research or in the clinic has been hampered by the complexity of the phenotype, which is determined by a myriad of genetic and epigenetic factors and a complex pathogenetic mechanism (Kalpatthi and Novelli, 2018). For example, even though VOEs are extremely disabling and the most common cause of hospitalization in SCD, self-reported pain scores remain the only indicator in humans, despite the fact that mouse models of SCD have allowed intravital imaging and characterization of the molecular events that lead to SCD (Sundd et al., 2019). This is a major limitation because self-reported pain is highly subjective and a notoriously poor biomarker. Indeed, both drug development and clinical care are hampered by the absence of objective, quantifiable biomarkers of SCD (Kalpatthi and Novelli, 2018).
Thus, it would be extremely valuable to develop new markers of acute pain for both clinical research and clinical care. Objective biomarkers would help validate self-reports and reduce mutual mistrust between patients and health care providers. In addition, there is a need for patient-centered, technology-based approaches that can capture distinctive changes in pain intensity and quality, such as the use of abstract animations, which have been shown to be less affected by age, literacy level, or language than visual analog score scales (Jonassaint et al., 2018a,b).
Patients are also afflicted by chronic pain. In the Pain in Sickle Cell Epidemiology Study (PiSCES), pain occurred daily in approximately half of study participants (McClish et al., 2017). Chronic pain is not typically included as an outcome in clinical research, yet it profoundly affects QOL. Thus, the development of a biomarker for chronic pain would be useful. Novel biomarkers under investigation include those based on brain imaging with functional MRI (Karafin et al., 2019).
SCD severity scores (Burke et al., 2016) and composite biomarker signatures (Du et al., 2018) are now being developed to address the limitations of existing VOE-based biomarkers.
To develop other endpoints for clinical research, investigators should focus on identifying biomarkers of specific pathogenic processes. The experience gained in the senicapoc trial indicates that hemolysis alone is not an adequate biomarker of efficacy for investigational drugs (Ataga et al., 2011). However, biomarkers that measure hyperadhesion, oxidant stress, or hemostatic activation could be employed to predict and measure response to drugs that specifically target those pathways (Kalpatthi and Novelli, 2018). In an effort to inform the field, ASH partnered with FDA to conduct work to identify clinical trial endpoints. The work, which was informed
by seven panels of clinicians, researchers, and patients, resulted in the publication of two consensus recommendations on (1) endpoints for patient-reported outcomes (PROs), pain, and the brain; and (2) endpoints for renal and cardiopulmonary, cure, and low-resource settings (Farrell et al., 2019).
Outcome Measures in SCD
There is an ongoing debate on clinically relevant endpoints for SCD research. The consensus is that the endpoints traditionally used in SCD clinical trials are limited and poorly represent the heterogeneity of the disease. Well-characterized biomarkers and relevant surrogate endpoints are scarce. While the validity of transcranial Doppler velocity to assess stroke risk and cerebral vasculopathy (Adams, 2007) and of tricuspid regurgitant jet velocity to assess mortality risk are undisputed, most other complications lack adequate biomarkers (Gladwin et al., 2004). VOE represents the most blatant example of a complication that has posed significant challenges for clinical trial design. Most studies have employed the duration and severity of VOE as clinical endpoints, yet both are difficult to operationalize.
One strategy to develop more valuable outcome measures has been to invest in developing tools for PROs. In particular, QOL measures and PROs that capture certain complications have not been adequately incorporated in clinical research. For instance, PRO instruments that include cognition, depressive symptoms, sexual dysfunction, and sleep disturbances have been lacking and are sorely needed. Other areas of development include measures that can be applied to younger and older populations and measures that capture the totality of acute and chronic pain. Examples of recommended PROs include the Brief Pain Inventory, the ASCQ-Me, and the Adult Sickle Cell Quality of Life Measurement Information System (ASCQ-Me) Quality of Care.2
HEALTH CARE DELIVERY POLICY
Once a new therapy has been developed, it must be provided to patients, which raises the next question: the most effective approaches for delivery. This is particularly important for new curative therapies, as these will typically involve different treatment regimens—a one-time treatment, for example, rather than daily dosing—and will likely be significantly more expensive than therapeutic drugs.
___________________
2 For more information on the tools, see http://www.ascq-me.org (accessed July 6, 2020) and https://www.mdanderson.org/documents/Departments-and-Divisions/Symptom-Research/BPI_UserGuide.pdf (accessed July 6, 2020).
Implications of Curative Therapy for Delivery System Innovation
Because traditional fee-for-service models of reimbursement provide little direct incentive for physicians to maximize quality or avoid wasteful spending, several recent reforms have attempted to strengthen physician incentives for quality and efficiency. Such reforms remain nascent in the SCD context, however. High-cost curative therapy may also pose unique challenges that could further stifle such delivery system reform innovations in SCD. This section considers these issues and potential solutions.
Patient-centered medical homes (PCMHs) are provider-centered care delivery models that encourage care coordination (AHRQ, n.d.), and they show promise for SCD care. Under this model, care is accessible, continuous, comprehensive, family-centered, coordinated, compassionate, and culturally effective (AAP, 2002; Raphael et al., 2013). Typically, care is coordinated by a personal physician or a provider in the community. Some evidence suggests that PCMHs improve outcomes for SCD patients. A study of 150 children in a large children’s hospital found that SCD children receiving PCMH had half the rate of ED visits (incident rate ratio [IRR] = 0.51, 95% CI 0.33–0.78) and just more than half the rate of hospitalizations (IRR = 0.56, 95% CI 0.33–0.93) compared with children not receiving such care (Raphael et al., 2013). The study found that the comprehensive care component of the PCMH was the one that significantly reduced ED visits and hospitalizations. This suggests that PCMH may hold promise as a delivery system reform. Caution is nonetheless warranted. While the PCMH may prove beneficial in the pediatric context, it may not be a viable method for improving the delivery of medical care for adults (Ballas and Vichinsky, 2015). The PCMH is centered around having a personal provider who will coordinate care as the primary criterion. Given the well-known problems that arise in transitioning from pediatrics to adult care, the PCMH model may be harder to establish in the adult care setting.
Another prominent delivery system reform is the use of the accountable care organization (ACO), which is a partnership between a third-party payer and a set of providers, typically including a primary care provider, specialists, hospitals, rehabilitation centers, and long-term care facilities, that seeks to provide coordinated care (CMS, 2019). ACOs receive a fixed base payment from the insurer plus a share in the savings if they achieve cost and quality targets for their patients. The goal is to encourage high-quality care and avoid unnecessary costs. One example is New York City’s Kings County ACO, which identified advanced SCD as a priority subpopulation due to the high number of readmissions (Stine et al., 2017). In response, the primary care and hematology departments partnered to create a multidisciplinary high-risk clinic to deal with advanced SCD patients. Systematic data on the performance of such ACOs in SCD care could better inform an overall delivery reform strategy.
Bundled payments are a third prominent reform type, designed to achieve lower costs, higher quality, and better health (Oyeku and Faro, 2017). Also known as “episode-based payments,” bundled payments are designed to encourage high-value care by encouraging providers to improve coordination, efficiency, and care quality and outcomes (NEJM Catalyst, 2018). Under bundled payments, the total health care costs for an episode are prospectively determined, and providers receive no marginal compensation for any services within the bundle. Thus, providers take a loss when the cost exceeds the prospective bundled payment level, but they can share in the savings if they achieve costs below the target (conditional on meeting quality standards). While there are examples of SCD bundles (e.g., the Cleveland Clinic Children’s SCD discharge bundle), they are relatively scarce. The Center for Medicare & Medicaid Innovation has developed more than 50 different payment and delivery models since its establishment via the Patient Protection and Affordable Care Act of 2010, but individuals with rare conditions such as SCD have been left behind.3
Delivery system innovation in SCD has not been rapid, and recent curative therapies pose additional challenges. If gene therapies or other curative therapies involve high up-front costs for health care providers (e.g., as in chimeric antigen receptor T-cell therapy for cancer treatment), in which hospitals must pay in advance to acquire the drug and then wait for reimbursement, it may discourage health systems and other provider organizations from delivery system innovation that might benefit (and thus attract) more SCD patients. A solution might be helping providers finance the up-front costs of high-cost therapies and care. Public and private payers might be tapped as a source of financing, especially since delays in reimbursement create the need for financing.
Curative therapies also pose a challenge for bundled payments. Incorporating these in a bundled payment would likely discourage their use because the fixed payment may be insufficient. On the other hand, excluding them might encourage overuse because the bundled payment itself penalizes overuse. Criteria for use of these new therapies would help address this problem, for example, reducing the number of “marginal” cases where patients may or may not benefit would help mitigate overuse, even if the curative therapies are not bundled.
It may be tempting to believe that curative therapy would eliminate the need for intensive, coordinated care, but this may not be the case. “Cured” individuals may still experience pain throughout their lives and may also require genetic counseling because they could pass the mutation to future generations. Experience with HSCT patients suggests that a fraction of
___________________
3 For a list of innovation models for payment and service delivery, see https://innovation.cms.gov/innovation-models#views=models (accessed July 8, 2020).
them continue to have chronic pain (Darbari et al., 2019). SCD patients may also continue to suffer the consequences of earlier organ damage.
Treatment innovation could even increase the need for specialized care. Curative therapies might be administered primarily or even solely by SCD specialists. If so, then access to specialists will limit therapy availability. Among commercially insured SCD patients, less than half see a specialist in a given year; that rate is less than one-sixth for Medicaid patients (Dampier et al., 2017). Unless access to specialists is particularly well correlated with eligibility for a therapy, barriers to specialist care will slow the diffusion of novel curative therapies. The solution must lie in either expanding specialist access or establishing protocols for nonspecialists—perhaps with some additional training, specialist support, or other resources.
Shared Decision Making
There are various reasons for believing that the best approach to making decisions about SCD care will be one in which patients and clinicians work together. For example, Ross et al. (2016) present qualitative research indicating that patients prefer collaborative decision-making processes over decisions made solely by clinicians. Patients also believe that clinicians should consider their personal preferences for care, listen to their perspective, and provide information about complications, long-term outcomes, side effects, and other relevant factors. The desire for SDM is likely to be even more acute in the context of curative therapy. “Cures” offer the chance of a substantial and durable clinical benefit, but they generally come with considerable uncertainty surrounding the degree of benefit and long-term side effects, and both the adverse effects and benefits are hard to gauge. The optimal clinical decision will depend at least in part on the patient’s tolerance for uncertainty and ambiguity. All of these factors suggest that an SDM approach is best.
Existing studies on SDM in the SCD context provide a basis for specific research relative to curative SCD therapy. Khemani et al. (2018) conducted qualitative research designed to elicit the factors influencing the choice of HSCT. Identifying the key decision factors helps facilitate more productive conversations between providers and patients about treatment choice. Crosby et al. (2015) set forth a set of six strategies designed to facilitate SDM in the context of weighing the benefits and side effects of HU treatment. Finally, an ongoing Patient-Centered Outcomes Research Institute study is comparing alternative approaches to facilitating SDM concerning treatment options for pediatric SCD patients (PCORI, 2019). The launch of an innovative curative therapy should be accompanied by careful research and guidance on strategies for SDM.
REIMBURSEMENT POLICY
Decisions about new SCD therapies will also depend strongly on various economic factors, such as the price of treatment, cost–benefit considerations, and, in particular, reimbursement policies. While it is impossible to offer any specifics about the economic future for novel SCD therapies, other areas of medicine offer a sense of the general economic considerations that will shape the uptake of these new SCD treatments and cures.
It is difficult to predict how curative therapies for SCD will be priced, but the relevant factors will include the size of the eligible patient population and the expected take-up, the expected rate of treatment response, and the extent of irreversible sequelae (e.g., organ damage). If the expected take-up is low, the budget impact of even an expensive therapy might be relatively limited for an individual payer; this would mitigate pushback on price. In addition, the expected clinical value will play a role in the price negotiations between pharmaceutical manufacturers and third-party payers. There remains considerable uncertainty around all of these factors.
This caveat notwithstanding, there is some insight from studying the lifetime cost of caring for SCD patients using chronic therapy. A “perfect” cure would be worth at least this much to third-party payers. For example, the average lifetime cost of treating a person with hepatitis C virus (HCV) is estimated to be $64,490, with higher costs for individuals with longer-than-average life expectancies (Razavi et al., 2013). Sofosbuvir (Sovaldi), which cured HCV in more than 90 percent of patients (Cholongitas and Papatheodoridis, 2014), launched with a list price of about $84,000, although payers likely paid somewhat less after rebates (particularly after competitors arrived) (ICER, 2015; Pollack, 2015; U.S. Senate Committee on Finance, 2015). Partially effective cures could be valued similarly, on a pro-rata basis. Note that these are minimum values because they disregard the value of health gains to patients, caregivers, employers, and other stakeholders. Nonetheless, they provide a fixed point for thinking about the potential cost and budget impact of cures at a time when details about their clinical benefits remain scarce.
Kauf et al. (2009) estimated the total lifetime cost of care for an SCD Medicaid patient to be more than $450,000. This is a conservative estimate because commercially insured patients cost more than Medicaid patients. Thus, in the current pricing environment, a perfect cure might cost upward of $450,000 per patient; $1 billion would treat just more than 2,000 patients, a fraction of those with SCD. Given these numbers, it is difficult to imagine that a large subset of patients would receive curative therapy without significant changes to the structure of pricing contracts or price levels. It is similarly unlikely that third-party payers will make investments of this size and scope without an evidence base assessing the value
of therapy—especially in light of growing willingness by U.S. payers to price according to measured effectiveness (CVSHealth, 2018). For curative therapies to be widely adopted, strategies will be required to stimulate the development of evidence on value and new approaches to payment.
Aligning Endpoints for Approval and Reimbursement
It is well understood that regulatory agencies and third-party payers differ in their incentives and demands for evidence on efficacy, safety, and value (Bognar et al., 2017). For example, FDA does not consider cost or value for money in its approval decisions. U.S. third-party payers, on the other hand, are increasingly considering value in their reimbursement decisions. Within the past year, the pharmacy benefit manager CVS Caremark proposed allowing clients to exclude pharmaceuticals that have a launch price that is higher than $100,000 per quality-adjusted life-year (QALY) gained due to the therapy (CVSHealth, 2018). Other payers are routinely using cost-per-QALY evidence in their price negotiations. A survey of 422 formulary decision makers from U.S. payers found that 45 percent were likely or extremely likely to request a rebate to align a drug’s net price with the recommended value-based price calculated by the Institute for Clinical and Economic Review (ICER) and that around 59 percent of payers have used an ICER report as a basis for clinical and economic outcomes thresholds when creating an outcomes-based contract (ICON, 2018).
Clinical trials will always feature endpoints likely to satisfy regulators. However, patients may lose out on access to new therapies if those endpoints do not also permit third-party payers to assess value. The dominant method for value assessment today is measuring cost per QALYs gained or some variant of that, such as cost per life-year saved or healthy life-year gained. This is not to suggest that the cost per QALY is or should be the only criterion for reimbursement. Rather, payers are more likely to cover a new therapy generously if there is evidence of value for the money.
The extant cost-effectiveness literature on SCD therapies is somewhat sparse; a search of PubMed generated only 62 results, and very few of them used QALYs to measure benefit. Spackman et al. (2014) did so, reporting the cost per QALY gained by preoperative transfusion. Other studies have used alternatives to the QALY as a measure of benefit. For example, Panepinto et al. (2000) estimated the cost per additional life-year saved by universal SCD screening compared with targeted screening. Other studies have reported cost per healthy life-year gained (Cunningham-Myrie et al., 2015; McGann et al., 2015).
Controversy persists around whether QALYs are the best way to measure health benefits (Lakdawalla et al., 2018). However, the increasing adoption of cost-per-QALY criteria by U.S. payers suggests that evidence
on QALYs can ease the path toward access for patients. A critical step will be to map clinical endpoints to “health utility levels.” Very briefly, each health state (e.g., the number of pain crises per month) is enumerated and then valued relative to a perfectly healthy life-year. Spending 1 year in a state that is 80 percent as valuable as a perfectly healthy life-year is worth 0.8 QALYs, and so on. Innovators should anticipate the demand for studies on value by generating studies that map their clinical trial endpoints of interest to QALYs. Producing evidence on value more quickly will facilitate a more rapid uptake of new therapies (Lakdawalla et al., 2018).
There is a limited body of evidence estimating QALYs in the sickle cell context. A few studies have estimated health utility levels for SCD patients (Anie et al., 2002; McClish et al., 2017; Woods et al., 1997). The United Kingdom’s National Institute for Health and Care Excellence (NICE) reviewed this literature and calculated that the weighted average QALY for SCD was 0.732 (i.e., 1 year spent with SCD is estimated to be worth 0.732 perfectly healthy life-years) (NICE, 2012). This study also extrapolated utility levels associated with two complications, ACS and stroke; because of the absence of SCD-specific data, these were based on estimates obtained in non-SCD patient populations.
The growing importance of QALYs, along with the relatively limited literature on QALY measurement in SCD, suggests the importance of generating data that can readily assess the value of new SCD therapies. In addition, clinical trial endpoints must align with the published methods for estimating QALYs. For example, an innovator may conduct a clinical trial showing that a new drug reduces the number of monthly pain crises. This permits an assessment of value only if studies exist that compute health utility levels and QALYs against monthly pain crises.
Value assessments include both estimated QALYs gained and the incremental costs of a new therapy. Cost estimates include the direct cost of a new intervention and of supportive care as well as the cost offsets associated with, for example, reduced health care use due to health benefits. The International Society for Pharmacoeconomics and Outcomes Research published guidelines in 2015 recommending that collecting cost data should be fully integrated into clinical trials (Ramsey et al., 2015). Including costs as a secondary endpoint in pivotal randomized clinical trials would expedite the collection of these data and the associated assessment of value.
Novel Payment Mechanisms for Curative Therapies
Lessons from Other Disease Areas
Curative therapies may provide substantial long-term benefits but with high up-front costs. Several noteworthy examples in other disease areas
provide insights into the kinds of access constraints that SCD cures may face and the potential for solutions.
Sovaldi, as noted earlier, cured HCV in more than 90 percent of patients (Cholongitas and Papatheodoridis, 2014). The treatment was initially priced at about $84,000 for a 12-week course in the United States. While the price fell in the months following launch, it remained relatively high (ICER, 2015; Pollack, 2015). Significantly, Sovaldi provided value for money (NICE, 2014). NICE deemed the United Kingdom price of about GBP 35,000 to be cost-effective (Hirschler, 2014; NICE, 2014). Despite the evidence on value, Sovaldi’s price multiplied by the number of HCV patients resulted in a substantial potential budget impact (Henry, 2018; Touchot and Flume, 2015). During the first 9 months of 2014, the total U.S. health care expenditures were $6.6 billion, making Sovaldi the highest-revenue drug in the country during that period (Schumock et al., 2015).
At the same time, reports emerged of payers denying coverage of Sovaldi or approving access only after fibrotic changes had been documented in the liver (Do et al., 2015). Several private insurers implemented stringent prior authorization systems to limit their spending on Sovaldi, to the point that 25 percent of patients prescribed it were initially denied access (Schumock et al., 2015), even though many did receive it later (Do et al., 2015).
Sovaldi presents a case of a likely cost-effective drug whose uptake was limited by its substantial budget impact. This raises the question of whether novel financing arrangements can blunt the problem of high up-front costs. One direct approach is an annuity pricing model, sometimes referred to as a “drug mortgage” (Sachs et al., 2018). For example, Bluebird Bio has developed a gene-replacement therapy for thalassemia that may feature a seven-figure up-front cost. Perhaps as a result, it has announced plans for an annuity payment model, in which third-party payers would pay on a 5-year installment plan, with the installments also partially tied to the continuing durable effectiveness of the drug (Walker, 2019). Other firms have also tied the price of cures to their effectiveness. Spark Therapeutics prices its gene therapy for childhood retinal dystrophy at $850,000. Given the uncertainty about its long-term effectiveness and its high price tag, the company has proposed an outcomes-based pricing contract that offers a partial refund if the patient’s eyesight deteriorates (Richards, 2019). Spark Therapeutics also proposed a payment option that, in addition to the outcomes-based rebates, spreads out payments over time. Because current government price-reporting requirements do not explicitly account for the possibility of these amortized payments, Spark Therapeutics has proposed this novel payment mechanism as a Centers for Medicare & Medicaid Services (CMS) demonstration project (Spark Therapeutics, 2018). The obstacles created by federal price-reporting requirements for novel payments are considered in more detail below. Another pricing approach is to negotiate a fixed total
cost that can help payers reduce the risk of unexpectedly large budget impacts. For example, Louisiana’s Medicaid program is experimenting with a “Netflix-style” pricing model: for a fixed monthly subscription fee, it receives a license for unrestricted use of AbbVie’s, Gilead’s, or Merck’s HCV treatments (Louisiana Department of Health, 2019). Medicaid programs are in a better position than private payers to avoid regulatory pitfalls, so they may present a potential test bed for novel payment mechanisms.
Finally, while this issue is not unique to curative therapies, prior authorization rules for new therapies might become a barrier to patient access, particularly if there is no published evidence of value. Prior authorization rules require physicians or hospitals to submit formal requests to payers before providing certain treatments (AMCP, 2012). This tool is implemented as a cost-cutting strategy; in principle, it limits an expensive treatment to patients deemed genuinely in need. However, in a recent survey of physicians by the American Medical Association, 47 percent of respondents reported that prior authorization, when required, often or always delayed access to necessary treatments; 91 percent reported that it significantly or somewhat negatively affected patient clinical outcomes, and 28 percent reported that it led to a serious adverse event (e.g., death, hospitalization, disability/permanent bodily damage, or other life-threatening event) (AMA, 2018). Prior authorization becomes more likely when the case for value is weaker; it is a tool for limiting use rather than allocating treatment to patients likely to receive demonstrable higher value from it.
Innovative Payment Models for Curative or High-Cost Therapies
The examples above touch on a few of the numerous possible alternative payment mechanisms for curative therapies.
Annuity-style payment models allow health plans to spread out payments for treatments over time through installments up to a contractual ceiling (Carr and Bradshaw, 2016). Annuity-style payments suit treatments that provide value for money but have exceptionally high budget impacts because of large patient populations or high up-front costs. Annuity models can also be tied to outcomes, as in the thalassemia example above. Performance-based risk-sharing (PBRS) agreements have been around since the early 2000s (Carlson et al., 2017). PBRS agreements tie payments to the real-world performance of the annuity model. Because payers and manufacturers share the risk, PBRS agreements provide a means for addressing uncertainty about long-term therapeutic benefits or side effects. A study by Carlson et al. (2017) identified around 62 PBRS agreements in the United States between 1993 and 2016, of which 68 percent were active or presumed active in 2016: 47 percent were for pharmaceutical products, 34 percent for devices, and 19 percent for diagnostics. PBRS agreements do
require a clinical endpoint that can be measured in real-world data; pain crises might be one example, although a wider range of options would help facilitate outcomes-based pricing in different treatment contexts.
Another example, which has been used in solid organ and stem cell transplantation, is reinsurance (Slocomb and Werner, 2017). The health plan can enter into a financial arrangement—with either a manufacturer or third-party reinsurer—to cap total spending at some agreed-upon maximum. Reinsurance from a manufacturer simply caps spending at an agreed-upon level. Third-party reinsurance offloads spending above the threshold to the third party.
A related alternative is risk pooling. Under this scheme, payers would pay a certain share of their beneficiaries’ premiums into a fund devoted to high-cost therapies (Slocomb and Werner, 2017). If therapy costs exceed a prespecified threshold, the payer would receive compensatory funds from the pool. The pool could be administered by a nonprofit third party, the government, or the reinsurer, and it could be supported partially by the government to ensure patient access to high-cost treatments.
Enabling Innovative Contracting Arrangements
While there are a handful of innovative payment models, the idea remains nascent in the U.S. marketplace. New data—and policy and regulatory reforms—could help accelerate adoption.
As noted above, real-world data on effectiveness can insure payers against nonresponse or declining effectiveness over time. This requires identifying measurable endpoints that reflect patient well-being as comprehensively as possible.
Manufacturers have also shown some hesitation to enter into novel payment mechanisms due to Medicaid’s “best-price rule” (Sachs et al., 2018), under which Medicaid is legally entitled to pay the lowest price observed in the entire national marketplace for a drug. The implications for outcomes-based pricing remain slightly hazy. For example, with a contract that offers a full refund to a payer for a therapy that fails to work, if that refund is triggered for one patient in the plan, would that immediately entitle Medicaid to a price of zero? CMS could clarify the issue without congressional action. The Medicaid best-price statute does not specify the unit of analysis for a best-price calculation, that is, it does not stipulate whether the price paid for an individual patient or dose serves as a possible “best price.” Thus, for an outcomes-based contract, CMS could specify that the best price should be calculated as the weighted average of the prices the manufacturer receives (Sachs et al., 2018).
Clarity concerning the Anti-Kickback Statute, which bars manufacturers from offering federal health care programs anything of value in an
attempt to induce the purchase of a therapy (HEAT, n.d.), would also help in the case of Medicaid and Medicare payments. In principle, an outcomes-based rebate to Medicaid or Medicare might be construed as a kickback. Survey evidence shows that 57 percent of payers and 83 percent of manufacturers viewed clarifying the Anti-Kickback Statute as a high-impact, urgent need (Duhig et al., 2018).
Finally, annuity-style payment models might be hampered by the short duration of insurance contracts. SCD cures may have benefits that last for, say, 30 years. A natural response would be to spread payments out over this entire timeframe to mitigate the budget impact as much as possible. However, 17 percent of insurance beneficiaries switch their plan each year (Cunningham and Kohn, 2000). It is not entirely clear how to deal with an annuity payment obligation after a patient switches insurers. One solution would be to facilitate longer-term insurance contracts, but that would require far-reaching changes to the U.S. health insurance system (Bhattacharya et al., 2013). Another sweeping reform would be to create a system in which obligations can be transferred from one payer to another. Basu (2015) has proposed that payers receive a valuable tradable asset when they pay for a curative therapy. The value of this asset would reflect the total value of the cure to Medicare and Medicaid. This effectively rewards the payer for some or all of the benefits of the cure that accrue after the beneficiary enters public health insurance. It also enables the government to compensate commercial payers for some of the benefits that commercial payers are providing to public insurers.
Creating incentives for long-term health investments ought to be viewed as an important, long-term goal of health policy, but it must be supplemented with practical short-term steps as well. Existing examples suggest that shortening the annuity payout period may be a simpler, albeit less comprehensive, solution in the short term.
CONCLUSIONS AND RECOMMENDATIONS
Conclusion 7-1: For many African Americans (the predominant population group affected by SCD), efforts to advance medical science are still marred by historical exploitation. Yet, many are also willing to participate in clinical trials, and, somewhat paradoxically, there is evidence that recruitment efforts lag for this population. Researchers must be especially vigilant in their adherence to standards and practices in the consent and participation of those with SCD in research. Recruitment efforts for trials need to be equitable, including the use of community-based organizations to recruit and support participants. Communication with potential participants should be geared toward clarity and
education, and, over the long term, research results should be conveyed to participants whether the trial has been successful or not. This is especially warranted as new therapies (e.g., genetic modification, stem cell transplant) become widely available to treat and potentially cure those with SCD.
Conclusion 7-2: Many new disease-modifying drugs for SCD will become available in the next decades. Biomarkers are needed to predict response, prioritize certain treatments over others, and guide combination therapy. Comparative effectiveness research is needed to guide health care/insurance coverage policies, particularly for very expensive biological agents.
Conclusion 7-3: Allogeneic transplant from a sibling-matched donor carries limited toxicity and a high success rate. When HLA-matched siblings are available, stem cell transplant should be discussed and offered in early childhood, before complications have arisen. As new drugs that may obviate the need for curative treatment become available, biomarker-based decision trees are urgently needed to determine the appropriate timing and suitability of stem cell transplant. Additionally, there is a need for research on the impact of stem cell transplantation on organ dysfunction in adults.
Conclusion 7-4: Stem cell transplant carries a high psychosocial impact. Holistic care that addresses the psychological, economic, social, and spiritual impact of transplantation is critical and should become the standard of care from the pre-transplantation period to the post-transplant long-term follow-up stage.
Conclusion 7-5: Gene therapy advances and clinical trials are proceeding at a fast pace. Education and patient-facing materials on the risks and benefits of gene therapy are urgently needed, as is research to elucidate the long-term implications of gene therapy.
Conclusion 7-6: The current delivery and payment system could serve as an impediment to diffusion of and access to therapeutic innovations. There is a need for delivery system and payment reforms, including training of SCD specialists to facilitate the diffusion of new, curative, and other SCD therapies.
REFERENCES
AAP (American Academy of Pediatrics). 2002. The medical home. Pediatrics 110(1 Pt 1):184–186.
Acharya, K., C. W. Lang, and L. F. Ross. 2009. A pilot study to explore knowledge, attitudes, and beliefs about sickle cell trait and disease. Journal of the National Medical Association 101(11):1163–1172.
Adams, R. J. 2007. Hydroxyurea lowers TCD—and also stroke? Blood 110 (3):789–790.
AHRQ (Agency for Healthcare Research and Quality). n.d. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh (accessed January 10, 2020).
Allan, D., A. R. Limbrick, P. Thomas, and M. P. Westerman. 1982. Release of spectrin-free spicules on reoxygenation of sickled erythrocytes. Nature 295(5850):612–613.
AMA (American Medical Association). 2018. 2018 AMA prior authorization (PA) physician survey. https://www.ama-assn.org/system/files/2019-02/prior-auth-2018.pdf (accessed January 9, 2020).
AMCP (Academy of Managed Care Pharmacy). 2012. Prior authorization. http://www.amcp.org/prior_authorization (accessed April 13, 2020).
Anie, K. A., A. Steptoe, and D. H. Bevan. 2002. Sickle cell disease: Pain, coping and quality of life in a study of adults in the UK. British Journal of Health Psychology 7(Part 3):331–344.
Aslan, M., T. M. Ryan, B. Adler, T. M. Townes, D. A. Parks, J. A. Thompson, A. Tousson, M. T. Gladwin, R. P. Patel, M. M. Tarpey, I. Batinic-Haberle, C. R. White, and B. A. Freeman. 2001. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proceedings of the National Academy of Sciences 98(26):15215–15220.
ASRM (American Society for Reproductive Medicine). 2013. Fertility preservation in patients undergoing gonadotoxic therapy or gonadectomy: A committee opinion. Fertility and Sterility 100(5):1214–1223.
Ataga, K. I., and P. C. Desai. 2018. Advances in new drug therapies for the management of sickle cell disease. Expert Opinion on Orphan Drugs 6(5):329–343.
Ataga, K. I., and N. S. Key. 2007. Hypercoagulability in sickle cell disease: New approaches to an old problem. Hematology: American Society of Hematology—Education Program 91–96.
Ataga, K. I., C. G. Moore, C. A. Hillery, S. Jones, H. C. Whinna, D. Strayhorn, C. Sohier, A. Hinderliter, L. V. Parise, and E. P. Orringer. 2008. Coagulation activation and inflammation in sickle cell disease-associated pulmonary hypertension. Haematologica 93(1):20–26.
Ataga, K. I., M. Reid, S. K Ballas, Z. Yasin, C. Bigelow, L. S. James, W. R. Smith, F. Galacteros, A. Kutlar, J. H. Hull, J. W. Stocker, and the ICA-17043-10 study investigators. 2011. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: A Phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043). British Journal of Haematology 153(1):92–104.
Ataga, K. I., A. Kutlar, J. Kanter, D. Liles, R. Cancado, J. Friedrisch, T. H. Guthrie, J. Knight-Madden, O. A. Alvarez, V. R. Gordeuk, S. Gualandro, M. P. Colella, W. R. Smith, S. A. Rollins, J. W. Stocker, and R. P. Rother. 2017. Crizanlizumab for the prevention of pain crises in sickle cell disease. New England Journal of Medicine 376(5):429–439.
Bakshi, N., C. B. Sinha, D. Ross, K. Khemani, G. Loewenstein, and L. Krishnamurti. 2017. Proponent or collaborative: Physician perspectives and approaches to disease modifying therapies in sickle cell disease. PLOS ONE 12(7):e0178413.
Ballas, S. K., and E. P. Vichinsky. 2015. Is the medical home for adult patients with sickle cell disease a reality or an illusion? Hemoglobin 39(2):130–133.
Ballas, S. K., M. R. Kesen, M. F. Goldberg, G. A. Lutty, C. Dampier, I. Osunkwo, W. C. Wang, C. Hoppe, W. Hagar, D. S. Darbari, and P. Malik. 2012. Beyond the definitions of the phenotypic complications of sickle cell disease: An update on management. Scientific World Journal 2012:949535.
Basu, A. 2015. Financing cures in the United States. Expert Review of Pharmacoeconomics and Outcomes Research 15(1):1–4.
Belcher, J. D., C. Chen, J. Nguyen, L. Milbauer, F. Abdulla, A. I. Alayash, A. Smith, K. A. Nath, R. P. Hebbel, and G. M. Vercellotti. 2014. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123(3):377–390.
Bernaudin, F., G. Socie, M. Kuentz, S. Chevret, M. Duval, Y. Bertrand, J. P. Vannier, K. Yakouben, I. Thuret, P. Bordigoni, A. Fischer, P. Lutz, J. L. Stephan, N. Dhedin, E. Plouvier, G. Margueritte, D. Bories, S. Verlhac, H. Esperou, L. Coic, J. P. Vernant, E. Gluckman, and SFGM-TC. 2007. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 110(7):2749–2756.
Bernaudin, F., J. H. Dalle, D. Bories, R. Peffault de Latour, M. Robin, Y. Bertrand, C. Pondarre, J. P. Vannier, B. Neven, M. Kuentz, S. Maury, P. Lutz, C. Paillard, K. Yakouben, I. Thuret, C. Galambrun, N. Dhedin, C. Jubert, P. Rohrlich, J. O. Bay, F. Suarez, N. Raus, J. P. Vernant, E. Gluckman, C. Poirot, G. Socié, and Société Francaise de Greffe de Moelle et de Thérapie Cellulaire. 2019. Long-term event-free survival, chimerism and fertility outcomes in 234 patients with sickle-cell anemia younger than 30 years after myeloablative conditioning and matched-sibling transplantation in France. Haematologica 105(1):91–101.
Bhattacharya, J., A. Chandra, M. Chernew, D. Goldman, A. Jena, D. Lakdawalla, A. Malani, and T. Philipson. 2013. Best of both worlds: Uniting universal coverage and personal choice in health care. https://www.aei.org/wp-content/uploads/2014/09/-best-of-bothworlds-uniting-universal-coverage-and-personal-choice-in-health-care_105214167938.pdf (accessed January 9, 2020).
Blake, A., V. Asnani, R. R. Leger, J. Harris, V. Odesina, D. L. Hemmings, D. A. Morris, J. Knight-Madden, L. Wagner, and M. R. Asnani. 2018. Stigma and illness uncertainty: Adding to the burden of sickle cell disease. Hematology 23(2):122–130.
Bognar, K., J. A. Romley, J. P. Bae, J. Murray, J. W. Chou, and D. N. Lakdawalla. 2017. The role of imperfect surrogate endpoint information in drug approval and reimbursement decisions. Journal of Health Economics 51:1–12.
Bonham, V. L., C. Haywood, and V. N. Gamble. 2007. Sickle cell disease: The past, present and future social and ethical dilemmas. In B. Pace (ed.), Renaissance of sickle cell disease research in the genome era. London: Imperial College Press. Pp. 311–323.
Boulad, F., T. Shore, K. van Besien, C. Minniti, M. Barbu-Stevanovic, S. W. Fedus, F. Perna, J. Greenberg, D. Guarneri, V. Nandi, A. Mauguen, K. Yazdanbakhsh, M. Sadelain, and P. A. Shi. 2018. Safety and efficacy of plerixafor dose escalation for the mobilization of CD34+ hematopoietic progenitor cells in patients with sickle cell disease: Interim results. Haematologica 103(5):770–777.
Brodsky, R. A., L. Luznik, J. Bolanos-Meade, M. S. Leffell, R. J. Jones, and E. J. Fuchs. 2008. Reduced intensity HLA-haploidentical BMT with post transplantation cyclophosphamide in nonmalignant hematologic diseases. Bone Marrow Transplantation 42(8):523–527.
Brunson, A., A. Lei, A. S. Rosenberg, R. H. White, T. Keegan, and T. Wun. 2017. Increased incidence of VTE in sickle cell disease patients: Risk factors, recurrence and impact on mortality. British Journal Haematology 178(2):319–326.
Brunson, A., T. Keegan, A. Mahajan, R. White, and T. Wun. 2019. High incidence of venous thromboembolism recurrence in patients with sickle cell disease. American Journal of Hematology 94(8):862–870.
Bulgin, D., P. Tanabe, and C. Jenerette. 2018. Stigma of sickle cell disease: A systematic review. Issues in Mental Health Nursing 39(8):675–686.
Burke, L., J. C. Hobart, K. Fox, J. Lehrer-Graiwer, K. Bridges, M. Kraus, and E. Ramos. 2016. The 10-item sickle cell disease severity measure (SCDSM-10): A novel measure of daily SCD symptom severity developed to assess benefit of GBT440, an experimental HbS polymerization inhibitor. Blood 128(22):4760.
Cairo, M. S., J. A. Talano, T. B. Moore, Q. Shi, R. S. Weinberg, B. Grossman, and S. Shenoy. 2019. Familial haploidentical stem cell transplant in children and adolescents with high-risk sickle cell disease: A Phase 2 clinical trial. JAMA Pediatrics, December 9 [Epub ahead of print].
Cardenes, N., C. Corey, L. Geary, S. Jain, S. Zharikov, S. Barge, E. M. Novelli, and S. Shiva. 2014. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood 123(18):2864–2872.
Carlson, J. J., S. Chen, and L. P. Garrison, Jr. 2017. Performance-based risk-sharing arrangements: An updated international review. Pharmacoeconomics 35(10):1063–1072.
Carr, D. R., and S. E. Bradshaw. 2016. Gene therapies: The challenge of super-high-cost treatments and how to pay for them. Regenerative Medicine 11(4):381–393.
CDC (Centers for Disease Control and Prevention). 2015. U.S. Public Health Service syphilis study at Tuskegee: The Tuskegee timeline. https://www.cdc.gov/tuskegee/timeline.htm (accessed August 1, 2019).
Chang, J., J. T. Patton, A. Sarkar, B. Ernst, J. L. Magnani, and P. S. Frenette. 2010. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 116(10):1779–1786.
Charache, S., M. L. Terrin, R. D. Moore, G. J. Dover, F. B. Barton, S. V. Eckert, R. P. McMahon, D. R. Bonds, and Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. 1995. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. New England Journal of Medicine 332(20):1317–1322.
Chen, G., D. Zhang, T. A. Fuchs, D. Manwani, D. D. Wagner, and P. S. Frenette. 2014. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 123(24):3818–3827.
Children’s Hospital Los Angeles and Hope Pharmaceuticals. 2009. Safety and efficacy of sodium nitrite in sickle cell disease. https://clinicaltrials.gov/ct2/show/NCT01033227 (accessed January 9, 2020).
Cholongitas, E., and G. V. Papatheodoridis. 2014. Sofosbuvir: A novel oral agent for chronic hepatitis C. Annals of Gastroenterology 27(4):331–337.
CMS (Centers for Medicare & Medicaid Services). 2019. Accountable care organizations (ACOs): General information. https://innovation.cms.gov/initiatives/aco (accessed January 9, 2020).
Crary, S. E., and G. R. Buchanan. 2009. Vascular complications after splenectomy for hematologic disorders. Blood 114(14):2861–2868.
Crosby, L. E., L. M. Shook, R. E. Ware, and W. B. Brinkman. 2015. Shared decision making for hydroxyurea treatment initiation in children with sickle cell anemia. Pediatric Blood & Cancer 62(2):184–185.
Cunningham, P. J., and L. Kohn. 2000. Health plan switching: Choice or circumstance? Health Affairs 19(3):158–164.
Cunningham-Myrie, C., A. Abdulkadri, A. Waugh, S. Bortolusso Ali, L. G. King, J. Knight-Madden, and M. Reid. 2015. Hydroxyurea use in prevention of stroke recurrence in children with sickle cell disease in a developing country: A cost effectiveness analysis. Pediatric Blood & Cancer 62(10):1862–1864.
CVSHealth. 2018. Current and new approaches to making drugs more affordable. https://cvshealth.com/sites/default/files/cvs-health-current-and-new-approaches-to-making-drugs-more-affordable.pdf (accessed January 9, 2020).
Dallas, M. H., B. Triplett, D. R. Shook, C. Hartford, A. Srinivasan, J. Laver, R. Ware, and W. Leung. 2013. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biology of Blood and Marrow Transplantation 19(5):820–830.
Dampier, C., J. Kanter, R. Howard, I. Agoda, S. Wade, V. Noxon, and S. K. Ballas. 2017. Access to care for Medicaid and commercially-insured United States patients with sickle cell disease. Blood 130(Suppl 1):4660.
Darbari, D. S., J. Liljencrantz, A. Ikechi, S. Martin, M. C. Roderick, C. D. Fitzhugh, J. F. Tisdale, S. L. Thein, and M. Hsieh. 2019. Pain and opioid use after reversal of sickle cell disease following HLA-matched sibling haematopoietic stem cell transplant. British Journal of Haematology 184(4):690–693.
De Franceschi, L., M. D. Cappellini, and O. Olivieri. 2011. Thrombosis and sickle cell disease. Seminars in Thrombosis and Hemostasis 37(3):226–236.
de la Fuente, J., N. Dhedin, T. Koyama, F. Bernaudin, M. Kuentz, L. Karnik, G. Socie, K. A. Culos, R. A. Brodsky, M. R. DeBaun, and A. A. Kassim. 2019. Haploidentical bone marrow transplantation with post-transplantation cyclophosphamide plus thiotepa improves donor engraftment in patients with sickle cell anemia: Results of an international learning collaborative. Biology of Blood and Marrow Transplantation 25(6):1197–1209.
Demirci S., A. Leonard, J. J. Haro-Mora, N. Uchida, and J. F. Tisdale. 2019. CRISPR/Cas9 for sickle cell disease: Applications, future possibilities, and challenges. Advances in Experimental Medicine and Biology 1144:37–52.
Do, A., Y. Mittal, A. Liapakis, E. Cohen, H. Chau, C. Bertuccio, D. Sapir, J. Wright, C. Eggers, K. Drozd, M. Ciarleglio, Y. Deng, and J. K. Lim. 2015. Drug authorization for sofosbuvir/ledipasvir (Harvoni) for chronic HCV infection in a real-world cohort: A new barrier in the HCV care cascade. PLOS ONE 10(8):e0135645.
Du, M., S. Van Ness, V. Gordeuk, S. M. Nouraie, S. Nekhai, M. Gladwin, M. H. Steinberg, and P. Sebastiani. 2018. Biomarker signatures of sickle cell disease severity. Blood Cells, Molecules, and Diseases 72(Suppl 1):1–9.
Duhig, A. M., S. Saha, S. Smith, S. Kaufman, and J. Hughes. 2018. The current status of outcomes-based contracting for manufacturers and payers: An AMCP membership survey. Journal of Managed Care and Specialty Pharmacy 24(5):410–415.
Dutra, F. F., L. S. Alves, D. Rodrigues, P. L. Fernandez, R. B. de Oliveira, D. T. Golenbock, D. S. Zamboni, and M. T. Bozza. 2014. Hemolysis-induced lethality involves inflammasome activation by heme. Proceedings of the National Academy of Sciences 111(39):E4110–E4118.
Eapen, M., R. Brazauskas, M. C. Walters, F. Bernaudin, K. Bo-Subait, C. D. Fitzhugh, J. S. Hankins, J. Kanter, J. J. Meerpohl, J. Bolanos-Meade, J. A. Panepinto, D. Rondelli, S. Shenoy, J. Williamson, T. L. Woolford, E. Gluckman, J. E. Wagner, and J. F. Tisdale. 2019. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: A retrospective multicentre, cohort study. Lancet Haematology 6(11):e585–e596.
Esrick, E. B., J. P. Manis, H. Daley, C. Baricordi, H. Trebeden-Negre, F. J. Pierciey, M. Armant, S. Nikiforow, M. M. Heeney, W. B. London, L. Biasco, M. Asmal, D. A. Williams, and A. Biffi. 2018. Successful hematopoietic stem cell mobilization and apheresis collection using plerixafor alone in sickle cell patients. Blood Advances 2(19):2505–2512.
Estepp, J. H. 2018. Voxelotor (GBT440), a first-in-class hemoglobin oxygen-affinity modulator, has promising and reassuring preclinical and clinical data. American Journal of Hematology 93(3):326–329.
Evitt, N. H., S. Mascharak, and R. B. Altman. 2015. Human germline CRISPR-Cas modification: Toward a regulatory framework. American Journal of Bioethics 15(12):25–29.
Farrell, A. T., J. Panepinto, C. P. Carroll, D. S. Darbari, A. A. Desai, A. A. King, R. J. Adams, T. D. Barber, A. M. Brandow, M. R. DeBaun, M. J. Donahue, K. Gupta, J. S. Hankins, M. Kameka, F. J. Kirkham, H. Luksenburg, S. Miller, P. A. Oneal, D. C. Rees, R. Setse, V. A. Sheehan, J. Strouse, C. L. Stucky, E. M. Werner, J. C. Wood, and W. T. Zempsky. 2019. End points for sickle cell disease clinical trials: Patient-reported outcomes, pain, and the brain. Blood Advances 3(23):3982–4001.
FDA (U.S. Food and Drug Administration). 2017. FDA approved L-glutamine powder for the treatment of sickle cell disease. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approved-l-glutamine-powder-treatment-sickle-cell-disease (accessed January 9, 2020).
FDA. 2019. FDA approves crizanlizumab-tmca for sickle cell disease. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-crizanlizumab-tmca-sickle-cell-disease (accessed January 9, 2020).
Field, J. J., E. Majerus, V. R. Gordeuk, M. Gowhari, C. Hoppe, M. M. Heeney, M. Achebe, A. George, H. Chu, B. Sheehan, M. Puligandla, D. Neuberg, G. Lin, J. Linden, and D. G. Nathan. 2017. Randomized Phase 2 trial of regadenoson for treatment of acute vasoocclusive crises in sickle cell disease. Blood Advances 1(20):1645–1649.
Franck, P. F., E. M. Bevers, B. H. Lubin, P. Comfurius, D. T. Chiu, J. A. Op den Kamp, R. F. Zwaal, L. L. van Deenen, and B. Roelofsen. 1985. Uncoupling of the membrane skeleton from the lipid bilayer. The cause of accelerated phospholipid flip-flop leading to an enhanced procoagulant activity of sickled cells. Journal of Clinical Investigation 75(1):183–190.
George, A., S. Pushkaran, D. G. Konstantinidis, S. Koochaki, P. Malik, N. Mohandas, Y. Zheng, C. H. Joiner, and T. A. Kalfa. 2013. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 121(11):2099–2107.
Gilman, A. L., M. J. Eckrich, S. Epstein, C. Barnhart, M. Cannon, T. Fukes, M. Hyland, K. Shah, D. Grochowski, E. Champion, and A. Ivanova. 2017. Alternative donor hematopoietic stem cell transplantation for sickle cell disease. Blood Advances 1(16):1215–1223.
Gladwin, M. T., V. Sachdev, M. L. Jison, Y. Shizukuda, J. F. Plehn, K. Minter, B. Brown, W. A. Coles, J. S. Nichols, I. Ernst, L. A. Hunter, W. C. Blackwelder, A. N. Schechter, G. P. Rodgers, O. Castro, and F. P. Ognibene. 2004. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. New England Journal of Medicine 350(9):886–895.
Gladwin, M. T., G. J. Kato, D. Weiner, O. C. Onyekwere, C. Dampier, L. Hsu, R. W. Hagar, T. Howard, R. Nuss, M. M. Okam, C. K. Tremonti, B. Berman, A. Villella, L. Krishnamurti, S. Lanzkron, O. Castro, V. R. Gordeuk, W. A. Coles, M. Peters-Lawrence, J. Nichols, M. K. Hall, M. Hildesheim, W. C. Blackwelder, J. Baldassarre, J. F. Casella, and N. I. De. 2011. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: A randomized controlled trial. JAMA 305(9):893–902.
Gluckman, E., B. Cappelli, F. Bernaudin, M. Labopin, F. Volt, J. Carreras, B. Pinto Simoes, A. Ferster, S. Dupont, J. de la Fuente, J. H. Dalle, M. Zecca, M. C. Walters, L. Krishnamurti, M. Bhatia, K. Leung, G. Yanik, J. Kurtzberg, N. Dhedin, M. Kuentz, G. Michel, J. Apperley, P. Lutz, B. Neven, Y. Bertrand, J. P. Vannier, M. Ayas, M. Cavazzana, S. Matthes-Martin, V. Rocha, H. Elayoubi, C. Kenzey, P. Bader, F. Locatelli, A. Ruggeri, and M. Eapen. 2017. Sickle cell disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 129(11):1548–1556.
Gragert, L., M. Eapen, E. Williams, J. Freeman, S. Spellman, R. Baitty, R. Hartzman, J. D. Rizzo, M. Horowitz, D. Confer, and M. Maiers. 2014. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. New England Journal of Medicine 371(4):339–348.
Green, N. S., M. Bhatia, E. Y. Griffith, M. Qureshi, C. Briamonte, M. Savone, S. Sands, M. T. Lee, A. Lignelli, and A. M. Brickman. 2017. Enhanced long-term brain magnetic resonance imaging evaluation of children with sickle cell disease after hematopoietic cell transplantation. Biology of Blood and Marrow Transplantation 23(4):670–676.
Griffin, T. C., D. McIntire, and G. R. Buchanan. 1994. High-dose intravenous methylprednisolone therapy for pain in children and adolescents with sickle cell disease. New England Journal of Medicine 330(11):733–737.
Halsey, C., and I. A. Roberts. 2003. The role of hydroxyurea in sickle cell disease. British Journal of Haematology 120(2):177–186.
Han, J., S. L. Saraf, R. E. Molokie, and V. R. Gordeuk. 2019. Use of metformin in patients with sickle cell disease. American Journal of Hematology 94(1):E13–E15.
Haywood, C., Jr., M. C. Beach, S. Bediako, C. P. Carroll, L. Lattimer, D. Jarrett, and S. Lanzkron. 2011. Examining the characteristics and beliefs of hydroxyurea users and nonusers among adults with sickle cell disease. American Journal of Hematology 86(1):85–87.
Haywood, C., Jr., S. Lanzkron, S. Bediako, J. J. Strouse, J. Haythornthwaite, C. P. Carroll, M. Diener-West, G. Onojobi, M. C. Beach, and the IMPORT Investigators. 2014. Perceived discrimination, patient trust, and adherence to medical recommendations among persons with sickle cell disease. Journal of General Internal Medicine 29(12):1657–1662.
HEAT (Health Care Fraud Prevention and Enforcement Action Team). n.d. Comparison of the Anti-Kickback Statute and Stark Law. https://oig.hhs.gov/compliance/providercompliance-training/files/starkandakscharthandout508.pdf (accessed Januay 9, 2020).
Heeney, M. M., C. C. Hoppe, M. R. Abboud, B. Inusa, J. Kanter, B. Ogutu, P. B. Brown, L. E. Heath, J. A. Jakubowski, C. Zhou, D. Zamoryakhin, T. Agbenyega, R. Colombatti, H. M. Hassab, V. N. Nduba, J. N. Oyieko, N. Robitaille, C. I. Segbefia, and D. C. Rees. 2016. A multinational trial of prasugrel for sickle cell vaso-occlusive events. New England Journal of Medicine 374(7):625–635.
Henry, B. 2018. Drug pricing & challenges to hepatitis C treatment access. Journal of Health and Biomed Law 14:265–283.
Hirschler, B. 2014. UK cost body backs pricey Gilead hepatitis pill for some patients. Reuters, August 14. https://www.reuters.com/article/uk-health-hepatitis-gilead-sciences/uk-cost-body-backs-pricey-gilead-hepatitis-pill-for-some-patients-idUKKBN0GF05P20140815 (accessed April 15, 2020).
Horwitz, M. E., N. J. Chao, D. A. Rizzieri, G. D. Long, K. M. Sullivan, C. Gasparetto, J. P. Chute, A. Morris, C. McDonald, B. Waters-Pick, P. Stiff, S. Wease, A. Peled, D. Snyder, E. G. Cohen, H. Shoham, E. Landau, E. Friend, I. Peleg, D. Aschengrau, D. Yackoubov, J. Kurtzberg, and T. Peled. 2014. Umbilical cord blood expansion with nicotinamide provides long-term multilineage engraftment. Journal of Clinical Investigation 124(7):3121–3128.
Hsieh, M. M., and J. F. Tisdale. 2018. Hematopoietic stem cell mobilization with plerixafor in sickle cell disease. Haematologica 103(5):749–750.
Hsieh, M. M., E. M. Kang, C. D. Fitzhugh, M. B. Link, C. D. Bolan, R. Kurlander, R. W. Childs, G. P. Rodgers, J. D. Powell, and J. F. Tisdale. 2009. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. New England Journal of Medicine 361(24):2309–2317.
Hsieh, M. M., C. D. Fitzhugh, R. P. Weitzel, M. E. Link, W. A. Coles, X. Zhao, G. P. Rodgers, J. D. Powell, and J. F. Tisdale. 2014. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 312(1):48–56.
ICER (Institute for Clinical and Economic Review). 2015. New lower prices for Gilead hepatitis C drugs reach CTAF threshold for high health system value. https://icer-review.org/announcements/new-lower-prices-for-gilead-hepatitis-c-drugs-reach-ctaf-threshold-for-high-health-system-value (accessed January 10, 2020).
ICON. 2018. Industry perceptions and expectations: The role of ICER as an independent HTA organisation. https://www.iconplc.com/insights/value-based-healthcare/the-role-of-icer-as-an-independent-hta-organisation (accessed January 10, 2020).
Jinek, M., K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna, and E. Charpentier. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821.
Jonassaint, C. R., C. Kang, D. M. Abrams, J. J. Li, J. Mao, Y. Jia, Q. Long, M. Sanger, J. C. Jonassaint, L. De Castro, and N. Shah. 2018a. Understanding patterns and correlates of daily pain using the Sickle Cell Disease Mobile Application to Record Symptoms via Technology (SMART). British Journal of Haematology 183(2):306–308.
Jonassaint, C. R., N. Rao, A. Sciuto, G. E. Switzer, L. De Castro, G. J. Kato, J. C. Jonassaint, Z. Hammal, N. Shah, and A. Wasan. 2018b. Abstract animations for the communication and assessment of pain in adults: Cross-sectional feasibility study. Journal of Medical Internet Research 20(8):e10056.
Joseph, L., S. Manceau, C. Pondarré, A. Habibi, F. Bernaudin, S. Allali, M. De Montalembert, C. Chalas, J. B. Arlet, C. Jean, and V. Brousse. 2019. Effect of hydroxyurea exposure before puberty on sperm parameters in males with sickle cell disease. Blood 134(Suppl 1):889.
Joshi, S., B. N. Savani, E. J. Chow, M. H. Gilleece, J. Halter, D. A. Jacobsohn, J. Pidala, G. P. Quinn, J. Y. Cahn, A. A. Jakubowski, N. R. Kamani, H. M. Lazarus, J. D. Rizzo, H. C. Schouten, G. Socie, P. Stratton, M. L. Sorror, A. B. Warwick, J. R. Wingard, A. W. Loren, and N. S. Majhail. 2014. Clinical guide to fertility preservation in hematopoietic cell transplant recipients. Bone Marrow Transplant 49(4):477–484.
Kalpatthi, R., and E. M. Novelli. 2018. Measuring success: Utility of biomarkers in sickle cell disease clinical trials and care. Hematology: American Society of Hematology—Education Program 2018(1):482–492.
Kamani, N. R., M. C. Walters, S. Carter, V. Aquino, J. A. Brochstein, S. Chaudhury, M. Eapen, B. M. Freed, M. Grimley, J. E. Levine, B. Logan, T. Moore, J. Panepinto, S. Parikh, M. A. Pulsipher, J. Sande, K. R. Schultz, S. Spellman, and S. Shenoy. 2012. Unrelated donor cord blood transplantation for children with severe sickle cell disease: Results of one cohort from the Phase II study from the blood and marrow transplant clinical trials network (BMT CTN). Biology of Blood Marrow Transplantation 18(8):1265–1272.
Kanter, J., M. C. Walters, M. Hsieh, L. Krishnamurti, J. L. Kwiatkowski, R. Kamble, C. von Kalle, M. Joseney-Antoine, F. J. Pierciey, W. Shi, M. Asmal, A. A. Thompson, and J. F. Tisdale. 2017. Interim results from a Phase 1/2 clinical study of lentiglobin gene therapy for severe sickle cell disease. Blood 130(Suppl 1):527.
Kanter, J., A. Thompson, M. Mapara, J. Kwiatkowski, L. Krishnamurti, M. Schmidt, A. Miller, F. Pierciey, W. Huang, J. Ribeil, M. Walters, and J. Tisdale. 2019. Updated results from the HGB-206 study in patients with severe sickle cell disease treated under a revised protocol with lentiglobin gene therapy using plerixafor-mobilised haematopoietic stem cells. HemaSphere 3(S1):754–755.
Kapoor, S., J. A. Little, and L. H. Pecker. 2018. Advances in the treatment of sickle cell disease. Mayo Clinic Proceedings 93(12):1810–1824.
Karafin, M. S., G. Chen, N. J. Wandersee, A. M. Brandow, R. W. Hurley, P. Simpson, D. Ward, S. J. Li, and J. J. Field. 2019. Chronic pain in adults with sickle cell disease is associated with alterations in functional connectivity of the brain. PLOS ONE 14(5):e0216994.
Karnilowicz, W. 2011. Identity and psychological ownership in chronic illness and disease state. European Journal of Cancer Care (Engl) 20(2):276–282.
Kassim, A. A., and D. Sharma. 2017. Hematopoietic stem cell transplantation for sickle cell disease: The changing landscape. Hematology/Oncology Stem Cell Therapy 10(4):259–266.
Kato, G. 2015. A multi-center study of riociguat in patients with sickle cell diseases. https://clinicaltrials.gov/ct2/show/NCT02633397 (accessed January 10, 2020).
Kauf, T. L., T. D. Coates, L. Huazhi, N. Mody-Patel, and A. G. Hartzema. 2009. The cost of health care for children and adults with sickle cell disease. American Journal of Hematology 84(6):323–327.
Kaul, D. K., and R. P. Hebbel. 2000. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. Journal of Clinical Investigation 106(3):411–420.
Kaul, D. K., X. D. Liu, M. E. Fabry, and R. L. Nagel. 2000. Impaired nitric oxide-mediated vasodilation in transgenic sickle mouse. American Journal of Physiology Heart and Circulatory Physiology 278(6):H1799–H1806.
Khemani, K., D. Ross, C. Sinha, A. Haight, N. Bakshi, and L. Krishnamurti. 2018. Experiences and decision making in hematopoietic stem cell transplant in sickle cell disease: Patients’ and caregivers’ perspectives. Biology of Blood and Marrow Transplantation 24(5):1041–1048.
Kutlar, A., M. E. Reid, A. Inati, A. T. Taher, M. R. Abboud, A. El-Beshlawy, G. R. Buchanan, H. Smith, K. I. Ataga, S. P. Perrine, and R. G. Ghalie. 2013. A dose-escalation Phase IIa study of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. American Journal of Hematology 88(11):E255–E260.
Kutlar, A., J. Kanter, D. K. Liles, O. A. Alvarez, R. D. Cancado, J. R. Friedrisch, J. M. Knight-Madden, A. Bruederle, M. Shi, Z. Zhu, and K. I. Ataga. 2019. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. American Journal of Hematolgy 94(1):55–61.
Lagresle-Peyrou, C., F. Lefrere, E. Magrin, J. A. Ribeil, O. Romano, L. Weber, A. Magnani, H. Sadek, C. Plantier, A. Gabrion, B. Ternaux, T. Felix, C. Couzin, A. Stanislas, J. M. Treluyer, L. Lamhaut, L. Joseph, M. Delville, A. Miccio, I. Andre-Schmutz, and M. Cavazzana. 2018. Plerixafor enables safe, rapid, efficient mobilization of hematopoietic stem cells in sickle cell disease patients after exchange transfusion. Haematologica 103(5):778–786.
Lakdawalla, D. N., J. A. Doshi, L. P. Garrison, C. E. Phelps, A. Basu, and P. M. Danzon. 2018. Defining elements of value in health care—A health economics approach: An ISPOR special task force report. Value in Health 21(2):131–139.
Lebensburger, J. D., R. F. Sidonio, M. R. Debaun, M. M. Safford, T. H. Howard, and I. C. Scarinci. 2013. Exploring barriers and facilitators to clinical trial enrollment in the context of sickle cell anemia and hydroxyurea. Pediatric Blood & Cancer 60(8):1333–1337.
Lebensburger, J. D., L. M. Hilliard, L. E. Pair, R. Oster, T. H. Howard, and G. R. Cutter. 2015. Systematic review of interventional sickle cell trials registered in clinicaltrials.Gov. Clinical Trials (London, England) 12(6):575–583.
Liem, R. I., A. H. Cole, S. A. Pelligra, M. Mason, and A. A. Thompson. 2010. Parental attitudes toward research participation in pediatric sickle cell disease. Pediatric Blood & Cancer 55(1):129–133.
Limerick, E., and C. Fitzhugh. 2019. Choice of donor source and conditioning regimen for hematopoietic stem cell transplantation in sickle cell disease. Journal of Clinical Medicine 8(11):1997.
Long, K. A., S. B. Thomas, R. E. Grubs, E. A. Gettig, and L. Krishnamurti. 2011. Attitudes and beliefs of African-Americans toward genetics, genetic testing, and sickle cell disease education and awareness. Journal of Genetic Counseling 20(6):572–592.
Loren, A. W., E. Chow, D. A. Jacobsohn, M. Gilleece, J. Halter, S. Joshi, Z. Wang, K. A. Sobocinski, V. Gupta, G. A. Hale, D. I. Marks, E. A. Stadtmauer, J. Apperley, J. Y. Cahn, H. C. Schouten, H. M. Lazarus, B. N. Savani, P. L. McCarthy, A. A. Jakubowski, N. R. Kamani, B. Hayes-Lattin, R. T. Maziarz, A. B. Warwick, M. L. Sorror, B. J. Bolwell, G. Socie, J. R. Wingard, J. D. Rizzo, and N. S. Majhail. 2011. Pregnancy after hematopoietic cell transplantation: A report from the Late Effects Working Committee of the Center for International Blood and Marrow Transplant Research (CIBMTR). Biology of Blood and Marrow Transplantation 17(2):157–166.
Louisiana Department of Health. 2019. Department of Health receives three proposals for hepatitis C payment model. http://ldh.la.gov/index.cfm/newsroom/detail/5072 (accessed January 13, 2020).
Machado, R. F., R. J. Barst, N. A. Yovetich, K. L. Hassell, G. J. Kato, V. R. Gordeuk, J. S. R. Gibbs, J. A. Little, D. E. Schraufnagel, L. Krishnamurti, R. E. Girgis, C. R. Morris, E. B. Rosenzweig, D. B. Badesch, S. Lanzkron, O. Onyekwere, O. L. Castro, V. Sachdev, M. A. Waclawiw, R. Woolson, J. C. Goldsmith, M. T. Gladwin, and walk-PHaSST Investigators and Patients. 2011. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 118(4):855–864.
Malik, P., M. Grimley, C. T. Quinn, A. Shova, L. Courtney, C. Lutzko, T. A. Kalfa, O. Niss, P. A. Mehta, S. Chandra, E. Grassman, J. C. M. Van der Loo, S. Witting, D. Nordling, A. Shreshta, S. Felker, C. Terrell, L. Reeves, D. Pillis, L. Anastacia, F. D. Bushman, J. Knight-Madden, K. Kalinyak, S. M. Davies, and M. Asnani. 2018. Gene therapy for sickle cell anemia using a modified gamma globin lentivirus vector and reduced intensity conditioning transplant shows promising correction of the disease phenotype. Blood 132(Suppl 1):1021.
Matte, A., F. Zorzi, F. Mazzi, E. Federti, O. Olivieri, and L. De Franceschi. 2019. New therapeutic options for the treatment of sickle cell disease. Mediterranean Journal of Hematology and Infectious Diseases 11(1):e2019002.
McClish, D. K., W. R. Smith, J. L. Levenson, I. P. Aisiku, J. D. Roberts, S. D. Roseff, and V. E. Bovbjerg. 2017. Comorbidity, pain, utilization, and psychosocial outcomes in older versus younger sickle cell adults: The PiSCES project. Biomed Research International 2017:4070547.
McGann, P. T., and R. E. Ware. 2015. Hydroxyurea therapy for sickle cell anemia. Expert Opinion on Drug Safety 14(11):1749–1758.
McGann, P. T., S. D. Grosse, B. Santos, V. de Oliveira, L. Bernardino, N. J. Kassebaum, R. E. Ware, and G. E. Airewele. 2015. A cost-effectiveness analysis of a pilot neonatal screening program for sickle cell anemia in the Republic of Angola. Journal of Pediatrics 167(6):1314–1319.
Michaelis, M. C. 2019. The Sickle Cell Clinical Trials Network: One year in. The Hematologist 16(4):1.
Minneci, P. C., K. J. Deans, H. Zhi, P. S. Yuen, R. A. Star, S. M. Banks, A. N. Schechter, C. Natanson, M. T. Gladwin, and S. B. Solomon. 2005. Hemolysis-associated endothelial dysfunction mediated by accelerated NO inactivation by decompartmentalized oxyhemoglobin. Journal of Clinical Investigation 115(12):3409–3417.
Molokie, R., D. Lavelle, M. Gowhari, M. Pacini, L. Krauz, J. Hassan, V. Ibanez, M. A. Ruiz, K. P. Ng, P. Woost, T. Radivoyevitch, D. Pacelli, S. Fada, M. Rump, M. Hsieh, J. F. Tisdale, J. Jacobberger, M. Phelps, J. D. Engel, S. Saraf, L. L. Hsu, V. Gordeuk, J. DeSimone, and Y. Saunthararajah. 2017. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: A randomized Phase 1 study. PLOS Medicine 14(9):e1002382.
Morris, C. R., F. A. Kuypers, L. Lavrisha, M. Ansari, N. Sweeters, M. Stewart, G. Gildengorin, L. Neumayr, and E. P. Vichinsky. 2013. A randomized, placebo-controlled trial of arginine therapy for the treatment of children with sickle cell disease hospitalized with vaso-occlusive pain episodes. Haematologica 98(9):1375–1382.
Mpara, M., J. Tisdale, J. Kanter, J. Kwiatkowski, L. Krishnamurti, M. Schmidt, A. Miller, F. Pierciey, W. Shi, J. Ribeil, M. Asmal, A. Thompson, and M. C. Walters. 2019. Lentiglobin gene therapy in patients with sickle cell disease: Updated interim results from HGB-206. Biology of Blood and Marrow Transplantation 25(3):S64–S65.
Naik, R. P., M. B. Streiff, C. Haywood, Jr., J. A. Nelson, and S. Lanzkron. 2013. Venous thromboembolism in adults with sickle cell disease: A serious and under-recognized complication. American Journal of Medicine 126(5):443–449.
Naik, R. P., M. B. Streiff, C. Haywood, Jr., J. B. Segal, and S. Lanzkron. 2014. Venous thromboembolism incidence in the cooperative study of sickle cell disease. Journal of Thrombosis and Haemostasis 12(12):2010–2016.
NASEM (National Academies of Sciences, Engineering, and Medicine). 2017a. Human genome editing: Science, ethics, and governance. Washington, DC: The National Academies Press.
NASEM. 2017b. Pain management and the opioid epidemic: Balancing societal and individual benefits and risks of prescription opioid use. Washington, DC: The National Academies Press.
NEJM Catalyst. 2018. What are bundled payments? https://catalyst.nejm.org/what-are-bundled-payments (accessed January 13, 2020).
NICE (National Institute for Health and Care Excellence). 2012. Sickle cell acute painful episode: Management of an acute painful sickle cell episode in hospital. https://www.ncbi.nlm.nih.gov/books/NBK126761/pdf/Bookshelf_NBK126761.pdf (accessed March 6, 2020).
NICE. 2014. NICE consults on further draft guidance on the drug sofosbuvir (Sovaldi) for treating hepatitis C. https://www.nice.org.uk/news/press-and-media/NICE-consults-on-draft-guidance-on-the-drug-sofosbuvir-for-treating-hepatitis-C (accessed January 13, 2020).
Nienhuis, A. W., A. C. Nathwani, and A. M. Davidoff. 2017. Gene therapy for hemophilia. Molecular Therapies 25(5):1163–1167.
NIH (National Institutes of Health). 2018. NIH launches initiative to accelerate genetic therapies to cure sickle cell disease. https://www.nih.gov/news-events/news-releases/nih-launches-initiative-accelerate-genetic-therapies-cure-sickle-cell-disease (accessed March 6, 2020).
Niihara, Y., S. T. Miller, J. Kanter, S. Lanzkron, W. R. Smith, L. L. Hsu, V. R. Gordeuk, K. Viswanathan, S. Sarnaik, I. Osunkwo, E. Guillaume, S. Sadanandan, L. Sieger, J. L. Lasky, E. H. Panosyan, O. A. Blake, T. N. New, R. Bellevue, L. T. Tran, R. L. Razon, C. W. Stark, L. D. Neumayr, E. P. Vichinsky, and investigators of the Phase 3 Trial of L-glutamine in Sickle Cell Disease. 2018. A Phase 3 trial of L-glutamine in sickle cell disease. New England Journal of Medicine 379(3):226–235.
Noubouossie, D., N. S. Key, and K. I. Ataga. 2016. Coagulation abnormalities of sickle cell disease: Relationship with clinical outcomes and the effect of disease modifying therapies. Blood Reviews 30(4):245–256.
Novelli, E. M., C. Huynh, M. T. Gladwin, C. G. Moore, and M. V. Ragni. 2012. Pulmonary embolism in sickle cell disease: A case–control study. Journal of Thrombosis Haemostasis 10(5):760–766.
Omondi, N. A., S. E. Ferguson, N. S. Majhail, E. M. Denzen, G. R. Buchanan, A. E. Haight, R. J. Labotka, J. D. Rizzo, and E. A. Murphy. 2013. Barriers to hematopoietic cell transplantation clinical trial participation of African American and black youth with sickle cell disease and their parents. Journal of Pediatric Hematology/Oncology 35(4):289–298.
Oyeku, S. O., and E. Z. Faro. 2017. Rigorous and practical quality indicators in sickle cell disease care. Hematology: American Society of Hematology—Education Program 2017(1):418–422.
Panepinto, J. A., D. Magid, M. J. Rewers, and P. A. Lane. 2000. Universal versus targeted screening of infants for sickle cell disease: A cost-effectiveness analysis. Journal of Pediatrics 136(2):201–208.
PCORI (Patient-Centered Outcomes Research Institute). 2019. Comparing two ways to help parents of children with sickle cell disease decide on treatment with their doctors. https://www.pcori.org/research-results/2017/comparing-two-ways-help-parents-children-sickle-cell-disease-decide-treatment (accessed January 10, 2020).
Pecker, L. H., E. Salzberg, S. Chaturvedi, N. Zhao, M. S. Christianson, and S. M. Lanzkron. 2019. Anti-Mullerian hormone, a measure of ovarian reserve, is low in female subjects in the multi-center study of hydroxyurea. Blood 134(Suppl 1):890.
Persaud, A., and V. L. Bonham. 2018. The role of the health care provider in building trust between patients and precision medicine research programs. American Journal of Bioethics 18(4):26–28.
Persaud, A., S. Desine, K. Blizinsky, and V. L. Bonham. 2019. A CRISPR focus on attitudes and beliefs toward somatic genome editing from stakeholders within the sickle cell disease community. Genetics in Medicine 21(8):1726–1734.
Peters, M., B. E. Plaat, H. ten Cate, H. J. Wolters, R. S. Weening, and D. P. Brandjes. 1994. Enhanced thrombin generation in children with sickle cell disease. Thrombosis and Haemostasis 71(2):169–172.
Pfizer. 2019. Pfizer announces Phase 3 top-line results for rivipansel in patients with sickle cell disease experiencing a vaso-occlusive crisis. https://www.pfizer.com/news/press-release/press-release-detail/pfizer_announces_phase_3_top_line_results_for_rivipansel_in_patients_with_sickle_cell_disease_experiencing_a_vaso_occlusive_crisis (accessed March 6, 2020).
Platt, O. S., D. J. Brambilla, W. F. Rosse, P. F. Milner, O. Castro, M. H. Steinberg, and P. P. Klug. 1994. Mortality in sickle cell disease. Life expectancy and risk factors for early death. New England Journal of Medicine 330(23):1639–1644.
Pollack, A. 2015. Sales of Sovaldi, new Gilead hepatitis C drug, soar to $10.3 billion. https://www.nytimes.com/2015/02/04/business/sales-of-sovaldi-new-gilead-hepatitis-c-drugsoar-to-10-3-billion.html (accessed January 10, 2020).
Puri, A., C. Haywood, M. C. Beach, M. Guidera, S. Lanzkron, D. Valenzuela-Araujo, R. E. Rothman, and A. F. Dugas. 2016. Improving emergency providers’ attitudes toward sickle cell patients in pain. Journal of Pain and Symptom Management 51(3):628–632.
Ramsey, S. D., R. J. Willke, H. Glick, S. D. Reed, F. Augustovski, B. Jonsson, A. Briggs, and S. D. Sullivan. 2015. Cost-effectiveness analysis alongside clinical trials II—An ISPOR Good Research Practices Task Force report. Value in Health 18(2):161–172.
Raphael, J. L., T. L. Rattler, M. A. Kowalkowski, B. U. Mueller, T. P. Giordano, and D. C. Brousseau. 2013. Association of care in a medical home and health care utilization among children with sickle cell disease. Journal of the National Medical Association 105(2):157–165.
Razavi, H., A. C. Elkhoury, E. Elbasha, C. Estes, K. Pasini, T. Poynard, and R. Kumar. 2013. Chronic hepatitis C virus (HCV) disease burden and cost in the United States. Hepatology 57(6):2164–2170.
Ribeil, J. A., S. Hacein-Bey-Abina, E. Payen, A. Magnani, M. Semeraro, E. Magrin, L. Caccavelli, B. Neven, P. Bourget, W. El Nemer, P. Bartolucci, L. Weber, H. Puy, J. F. Meritet, D. Grevent, Y. Beuzard, S. Chretien, T. Lefebvre, R. W. Ross, O. Negre, G. Veres, L. Sandler, S. Soni, M. de Montalembert, S. Blanche, P. Leboulch, and M. Cavazzana. 2017. Gene therapy in a patient with sickle cell disease. New England Journal of Medicine 376(9):848–855.
Richards, S. E. 2019. When a treatment costs $450,000 or more, it had better work. The Atlantic, April 22.
Riley, T. R., A. Boss, D. McClain, and T. T. Riley. 2018. Review of medication therapy for the prevention of sickle cell crisis. Pharmacy and Therapeutics 43(7):417–437.
Ross, D., N. Bakshi, K. Khemani, C. Sinha, G. Loewenstein, and L. Krishnamurti. 2016. What are the expectations of patients in decision making process for disease modifying therapies for sickle cell disease: Do they care about shared decision making? Blood 128(22):5968.
Sachs, R., N. Bagley, and D. N. Lakdawalla. 2018. Innovative contracting for pharmaceuticals and Medicaid’s best-price rule. Journal of Health Politics, Policy and Law 43(1):5–18.
Saraf, S. L., A. L. Oh, P. R. Patel, Y. Jalundhwala, K. Sweiss, M. Koshy, S. Campbell-Lee, M. Gowhari, J. Hassan, D. Peace, J. G. Quigley, I. Khan, R. E. Molokie, L. L. Hsu, N. Mahmud, D. J. Levinson, A. S. Pickard, J. G. Garcia, V. R. Gordeuk, and D. Rondelli. 2016. Nonmyeloablative stem cell transplantation with alemtuzumab/low-dose irradiation to cure and improve the quality of life of adults with sickle cell disease. Biology of Blood and Marrow Transplantation 22(3):441–448.
Scharff, D. P., K. J. Mathews, P. Jackson, J. Hoffsuemmer, E. Martin, and D. Edwards. 2010. More than Tuskegee: Understanding mistrust about research participation. Journal of Health Care for the Poor and Underserved 21(3):879–897.
Schimmel, M., E. Nur, B. J. Biemond, G. J. van Mierlo, S. Solati, D. P. Brandjes, H. M. Otten, J. J. Schnog, S. Zeerleder, on behalf of the Curama Study Group. 2013. Nucleosomes and neutrophil activation in sickle cell disease painful crisis. Haematologica 98(11):1797–1803.
Schumock, G. T., E. C. Li, K. J. Suda, M. D. Wiest, J. Stubbings, L. M. Matusiak, R. J. Hunkler, and L. C. Vermeulen. 2015. National trends in prescription drug expenditures and projections for 2015. American Journal of Health-System Pharmacy 72(9):717–736.
Setty, B. N., S. G. Betal, J. Zhang, and M. J. Stuart. 2008. Heme induces endothelial tissue factor expression: Potential role in hemostatic activation in patients with hemolytic anemia. Journal of Thrombosis and Haemostasis 6(12):2202–2209.
Shapiro, B. S., L. J. Benjamin, R. Payne, and G. Heidrich. 1997. Sickle cell-related pain: Perceptions of medical practitioners. Journal of Pain Symptom Management 14(3):168–174.
Shearstone, J. R., O. Golonzhka, A. Chonkar, D. Tamang, J. H. van Duzer, S. S. Jones, and M. B. Jarpe. 2016. Chemical inhibition of histone deacetylases 1 and 2 induces fetal hemoglobin through activation of GATA2. PLOS ONE 11(4):e0153767.
Shenoy, S., J. Gaziev, E. Angelucci, A. King, M. Bhatia, A. Smith, D. Bresters, A. E. Haight, C. N. Duncan, J. de la Fuente, A. C. Dietz, K. S. Baker, M. A. Pulsipher, and M. C. Walters. 2018. Late effects screening guidelines after hematopoietic cell transplantation (HCT) for hemoglobinopathy: Consensus statement from the Second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects after Pediatric HCT. Biology of Blood and Marrow Transplantation 24(7):1313–1321.
Singer, S. T., and K. I. Ataga. 2008. Hypercoagulability in sickle cell disease and beta-thalassemia. Current Molecular Medicine 8(7):639–645.
Slocomb, T., and M. Werner. 2017. New payment and financing models for curative regenerative medicines. https://invivo.pharmaintelligence.informa.com/IV005132/New-Payment-And-Financing-Models-For-Curative-Regenerative-Medicines (accessed January 10, 2020).
Spackman, E., M. Sculpher, J. Howard, M. Malfroy, C. Llewelyn, L. Choo, R. Hodge, T. Johnson, D. C. Rees, K. Fijnvandraat, M. Kirby-Allen, S. Davies, and L. Williamson. 2014. Cost-effectiveness analysis of preoperative transfusion in patients with sickle cell disease using evidence from the taps trial. European Journal of Haematology 92(3):249–255.
Spark Therapeutics. 2018. Spark Therapeutics announces first-of-their-kind programs to improve patient access to Luxturna™ (voretigene neparvovecrzyl), a one-time gene therapy treatment. http://ir.sparktx.com/news-releases/news-release-details/spark-therapeutics-announces-first-their-kind-programs-improve (accessed January 10, 2020).
Stein, P. D., A. Beemath, F. A. Meyers, E. Skaf, and R. E. Olson. 2006. Deep venous thrombosis and pulmonary embolism in hospitalized patients with sickle cell disease. American Journal of Medicine 119(10).
Stein, R. 2019. The CRISPR revolution: A patient hopes gene-editing can help with pain of sickle cell disease. National Public Radio (WAMU 88.5). https://www.npr.org/sections/health-shots/2019/11/19/780510277/gene-edited-supercells-make-progress-in-fight-against-sickle-cell-disease (accessed January 30, 2020).
Steinberg, M. H. 1999. Management of sickle cell disease. New England Journal of Medicine 340(13):1021–1030.
Steinberg, M. H. 2002. Hydroxyurea treatment for sickle cell disease. Scientific World Journal 2:1706–1728.
Steinberg, M. H., and C. Brugnara. 2003. Pathophysiological-based approaches to treatment of sickle cell disease. Annual Review of Medicine 54:89–112.
Steinberg, M. H., W. F. McCarthy, O. Castro, S. K. Ballas, F. D. Armstrong, W. Smith, K. Ataga, P. Swerdlow, A. Kutlar, L. DeCastro, M. A. Waclawiw, Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia, and MSH Patients Follow-Up. 2010. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. American Journal of Hematology 85(6):403–408.
Stine, N., D. A. Chokshi, J. Knudsen, M. Cunningham, and R. Wilson. 2017. How America’s largest safety-net health system built a high performance Medicare ACO. Health Affairs, November 7. https://www.healthaffairs.org/do/10.1377/hblog20171106.437941/full (accessed April 13, 2020).
Strong, H., M. J. Mitchell, A. Goldstein-Leever, L. Shook, P. Malik, and L. E. Crosby. 2017. Patient perspectives on gene transfer therapy for sickle cell disease. Advances in Therapy 34(8):2007–2021.
Strouse, J. J., S. Lanzkron, M. C. Beach, C. Haywood, H. Park, C. Witkop, R. F. Wilson, E. B. Bass, and J. B. Segal. 2008. Hydroxyurea for sickle cell disease: A systematic review for efficacy and toxicity in children. Pediatrics 122(6):1332–1342.
Stuart, M. J., and R. L. Nagel. 2004. Sickle-cell disease. Lancet 364(9442):1343–1360.
Stuart, M. J., and B. N. Setty. 2001. Hemostatic alterations in sickle cell disease: Relationships to disease pathophysiology. Pediatric Pathology and Molecular Medicine 20(1):27–46.
Sundd, P., M. T. Gladwin, and E. M. Novelli. 2019. Pathophysiology of sickle cell disease. Annual Review of Pathology 14(1):263–292.
Tanhehco, Y. C., and M. Bhatia. 2019. Hematopoietic stem cell transplantation and cellular therapy in sickle cell disease: Where are we now? Current Opinion on Hematology 26(6):448–452.
Telen, M. J. 2007. Role of adhesion molecules and vascular endothelium in the pathogenesis of sickle cell disease. Hematology: American Society of Hematology—Education Program 2007:84–90.
Telen, M. J. 2016. Beyond hydroxyurea: New and old drugs in the pipeline for sickle cell disease. Blood 127(7):810–819.
Telen, M. J., T. Wun, T. L. McCavit, L. M. De Castro, L. Krishnamurti, S. Lanzkron, L. L. Hsu, W. R. Smith, S. Rhee, J. L. Magnani, and H. Thackray. 2015. Randomized Phase 2 study of GMI-1070 in SCD: Reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood 125(17):2656–2664.
Telen, M. J., P. Malik, and G. M. Vercellotti. 2019. Therapeutic strategies for sickle cell disease: Towards a multi-agent approach. Nature Reviews Drug Discovery 18(2):139–158.
Thompson, A. A. 2013. Ideal donors, imperfect results in sickle cell disease. Blood 122(6):858–859.
Tisdale, J. F., J. Kanter, M. Y. Mapara, J. L. Kwiatkowski, L. Krishnamurti, M. Schmidt, A. L. Miller, F. J. Pierciey, Jr., W. Shi, J.-A. Ribeil, M. Asmal, A. A. Thompson, and M. C. Walters. 2018. Current results of lentiglobin gene therapy in patients with severe sickle cell disease treated under a refined protocol in the Phase 1 HGB-206 study. Blood 132(Suppl 1):1026.
Tomer, A., S. Kasey, W. E. Connor, S. Clark, L. A. Harker, and J. R. Eckman. 2001. Reduction of pain episodes and prothrombotic activity in sickle cell disease by dietary n-3 fatty acids. Thrombosis and Haemostasis 85(6):966–974.
Touchot, N., and M. Flume. 2015. The payers’ perspective on gene therapies. Nature Biotechnology 33(9):902–904.
Treadwell, M. J., L. McClough, and E. Vichinsky. 2006. Using qualitative and quantitative strategies to evaluate knowledge and perceptions about sickle cell disease and sickle cell trait. Journal of the National Medical Association 98(5):704–710.
Tshilolo, L., G. Tomlinson, T. N. Williams, B. Santos, P. Olupot-Olupot, A. Lane, B. Aygun, S. E. Stuber, T. S. Latham, P. T. McGann, and R. E. Ware, for the REACH investigators. 2019. Hydroxyurea for children with sickle cell anemia in sub-Saharan Africa. New England Journal of Medicine 380(2):121–131.
U.S. Senate Committee on Finance. 2015. The price of Sovaldi and its impact on the U.S. health care system. https://www.finance.senate.gov/imo/media/doc/1%20The%20Price%20of%20Sovaldi%20and%20Its%20Impact%20on%20the%20U.S.%20Health%20Care%20System%20(Full%20Report).pdf (accessed March 6, 2020).
Vichinsky, E., C. C. Hoppe, K. I. Ataga, R. E. Ware, V. Nduba, A. El-Beshlawy, H. Hassab, M. M. Achebe, S. Alkindi, R. C. Brown, D. L. Diuguid, P. Telfer, D. A. Tsitsikas, A. Elghandour, V. R. Gordeuk, J. Kanter, M. R. Abboud, J. Lehrer-Graiwer, M. Tonda, A. Intondi, B. Tong, J. Howard, and Hope Trial investigators. 2019. A Phase 3 randomized trial of voxelotor in sickle cell disease. New England Journal of Medicine 381(6):509–519.
Villagra, J., S. Shiva, L. A. Hunter, R. F. Machado, M. T. Gladwin, and G. J. Kato. 2007. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood 110(6):2166–2172.
Vincent, L., D. Vang, J. Nguyen, M. Gupta, K. Luk, M. E. Ericson, D. A Simone, and K. Gupta. 2013. Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood 122(11):1853–1862.
Voskaridou, E., D. Christoulas, A. Bilalis, E. Plata, K. Varvagiannis, G. Stamatopoulos, K. Sinopoulou, A. Balassopoulou, D. Loukopoulos, and E. Terpos. 2010. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: Results of a 17-year, single-center trial (LASHS). Blood 115(12):2354–2363.
Wailoo, K. 2017. Sickle cell disease—A history of progress and peril. New England Journal of Medicine 376(9):805–807.
Walker, A. L., L. M. Gaydos, R. Farzan, L. De Castro, and C. Jonassaint. 2019. Social media discussions provide new insight about perceptions of hydroxyurea in the sickle cell community. American Journal of Hematology 94(5):E134–E136.
Walker, J. 2019. Biotech proposes paying for pricey drugs by installment. https://www.wsj.com/articles/biotech-proposes-paying-for-pricey-drugs-by-installment-11546952520 (accessed January 13, 2020).
Walters, M. C. 2005. Stem cell therapy for sickle cell disease: Transplantation and gene therapy. Hematology: American Society of Hematology—Education Program 2005:66–73.
Walters, M. C. 2015. Update of hematopoietic cell transplantation for sickle cell disease. Current Opinion in Hematology 22(3):227–233.
Walters, M. C., M. Patience, W. Leisenring, J. R. Eckman, J. P. Scott, W. C. Mentzer, S. C. Davies, K. Ohene-Frempong, F. Bernaudin, D. C. Matthews, R. Storb, and K. M. Sullivan. 1996. Bone marrow transplantation for sickle cell disease. New England Journal of Medicine 335(6):369–376.
Walters, M. C., R. Storb, M. Patience, W. Leisenring, T. Taylor, J. E. Sanders, G. E. Buchanan, Z. R. Rogers, P. Dinndorf, S. C. Davies, I. A. Roberts, R. Dickerhoff, A. M. Yeager, L. Hsu, J. Kurtzberg, K. Ohene-Frempong, N. Bunin, F. Bernaudin, W. Y. Wong, J. P. Scott, D. Margolis, E. Vichinsky, D. A. Wall, A. S. Wayne, C. Pegelow, R. Redding-Lallinger, J. Wiley, M. Klemperer, W. C. Mentzer, F. O. Smith, and K. M. Sullivan. 2000. Impact of bone marrow transplantation for symptomatic sickle cell disease: An interim report. Multicenter investigation of bone marrow transplantation for sickle cell disease. Blood 95(6):1918–1924.
Walters, M. C., M. Patience, W. Leisenring, Z. R. Rogers, V. M. Aquino, G. R. Buchanan, I. A. Roberts, A. M. Yeager, L. Hsu, T. Adamkiewicz, J. Kurtzberg, E. Vichinsky, B. Storer, R. Storb, K. M. Sullivan, and the Multicenter Investigation of Bone Marrow Transplantation for Sickle Cell Disease. 2001. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biology of Blood and Marrow Transplantation 7(12):665–673.
Walters, M. C., K. Hardy, S. Edwards, T. Adamkiewicz, J. Barkovich, F. Bernaudin, G. R. Buchanan, N. Bunin, R. Dickerhoff, R. Giller, P. R. Haut, J. Horan, L. L. Hsu, N. Kamani, J. E. Levine, D. Margolis, K. Ohene-Frempong, M. Patience, R. Redding-Lallinger, I. A. Roberts, Z. R. Rogers, J. E. Sanders, J. P. Scott, K. M. Sullivan, for the Multicenter Study of Bone Marrow Transplantation for Sickle Cell Disease. 2010. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biology of Blood and Marrow Transplantation 16(2):263–272.
Whelihan, M. F., M. Y. Lim, M. J. Mooberry, M. G. Piegore, A. Ilich, A. Wogu, J. Cai, D. M. Monroe, K. I. Ataga, K. G. Mann, and N. S. Key. 2016. Thrombin generation and cell-dependent hypercoagulability in sickle cell disease. Journal of Thrombosis and Haemostasis 14(10):1941–1952.
White, J., M. Lindgren, K. Liu, X. Gao, L. Jendeberg, and P. Hines. 2019. Sevuparin blocks sickle blood cell adhesion and sickle-leucocyte rolling on immobilized L-selectin in a dose dependent manner. British Journal of Haematology 184(5):873–876.
Woods, K., M. Miller, M. Johnson, A. Tracy, A. Kutlar, and C. Cassel. 1997. Functional status and well-being in adults with sickle cell disease. Journal of Clinical Outcomes Management 4(5):15–21.
Wun, T., T. Paglieroni, A. Rangaswami, P. H. Franklin, J. Welborn, A. Cheung, and F. Tablin. 1998. Platelet activation in patients with sickle cell disease. British Journal of Haematology 100(4):741–749.
Xue, C., X. Wang, Q. Qu, H. Qu, X. Fang, X. Sui, X. Liu, Y. Li, and Y. Jiang. 2019. Sexual dysfunction and abnormal androgen level after hematopoietic stem cell transplantation. Blood 134(Suppl 1):5691.


















































